本文要解读的文献如下

于2013年发表在nature methods杂志上,引用多达2115次。作为ATAC的开篇之作,在文章中详细介绍了ATAC的原理及应用,分为了以下几个部分
1. ATAC的实验方法
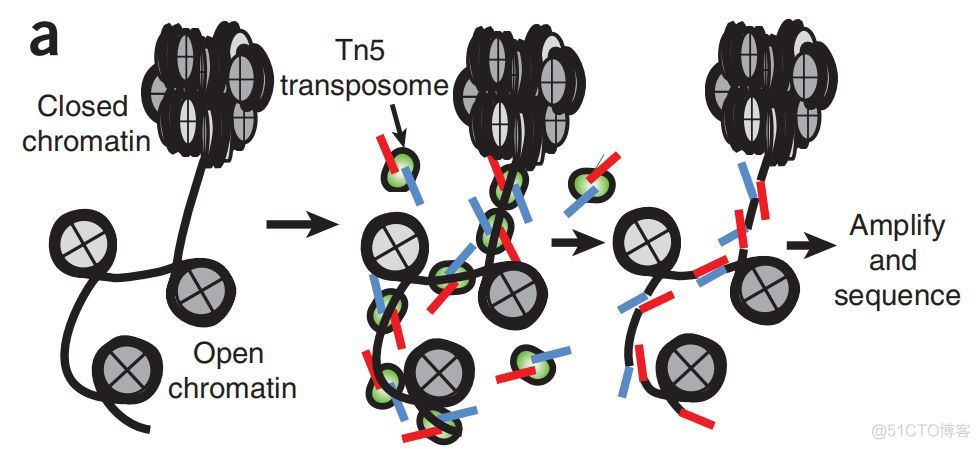
ATAC通过tn5转座酶来富集开放染色质区域的DNA序列,经PCR扩增后进行NGS测序,实验流程如下图所示
相比DNase-seq, FAIRE-seq, 该技术要求的细胞起始量低,而且文库构建耗时短,统计结果如下图所示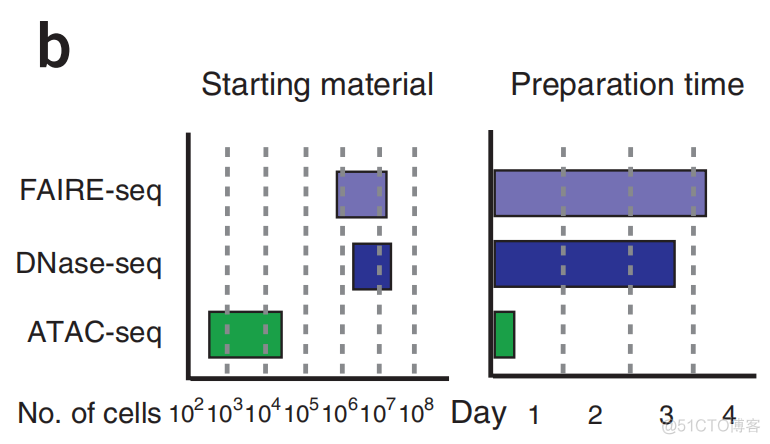
上图中的左侧描述了不同实验方法所需的细胞起始量,可以看到ATAC所需细胞最少,右侧描述了文库构建时间的长短,可以看到ATAC在1天之内就可以完成文库构建。
通过比较不同实验方法捕获构建的文库的一致性,发现ATAC重复性好,而且与其他实验方法的一致性高,对应的信号图如下所示
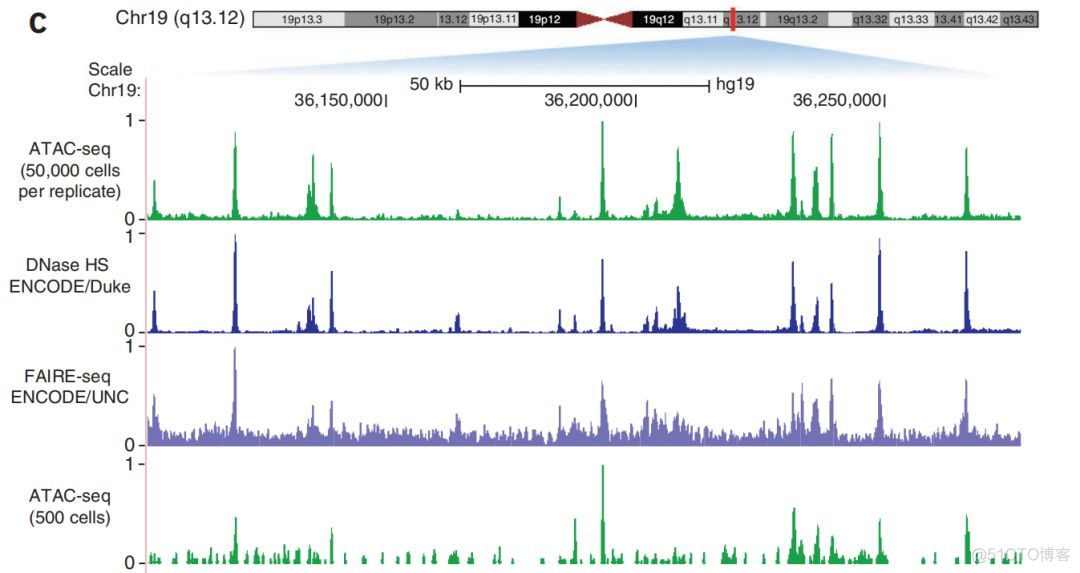
信号分布的峰型基本一致,相关性结果如下所示
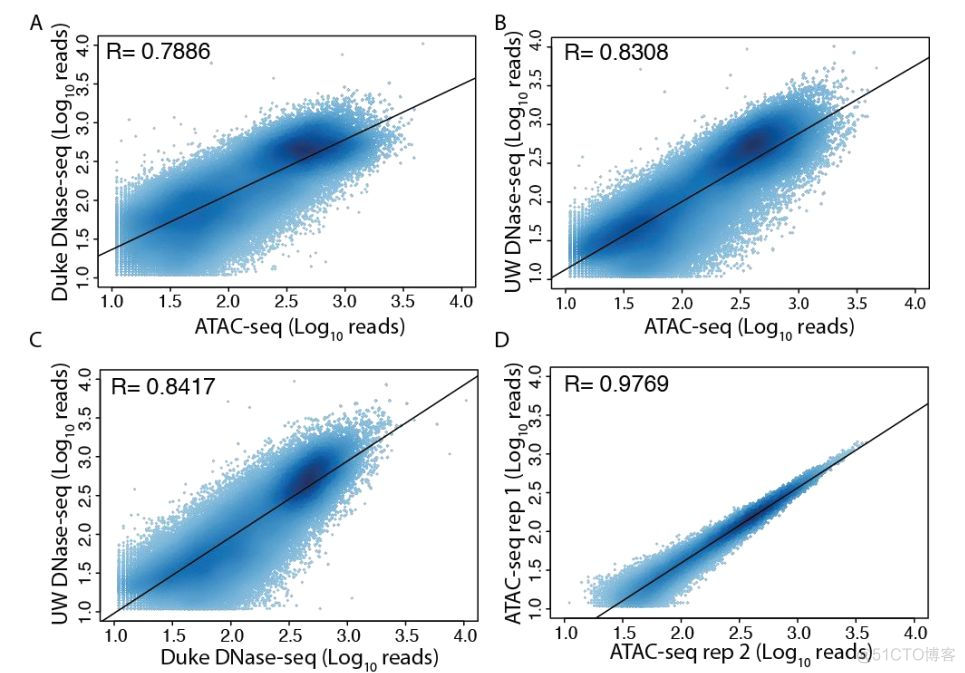
可以看到,相关系数都较高,两个ATAC文库的相关性则高达0.97。
2. ATAC的插入片段揭示了核小体的位置
通过对ATAC文库中插入片段的长度进行分析,观察到了一种很有意思的现象,示例如下
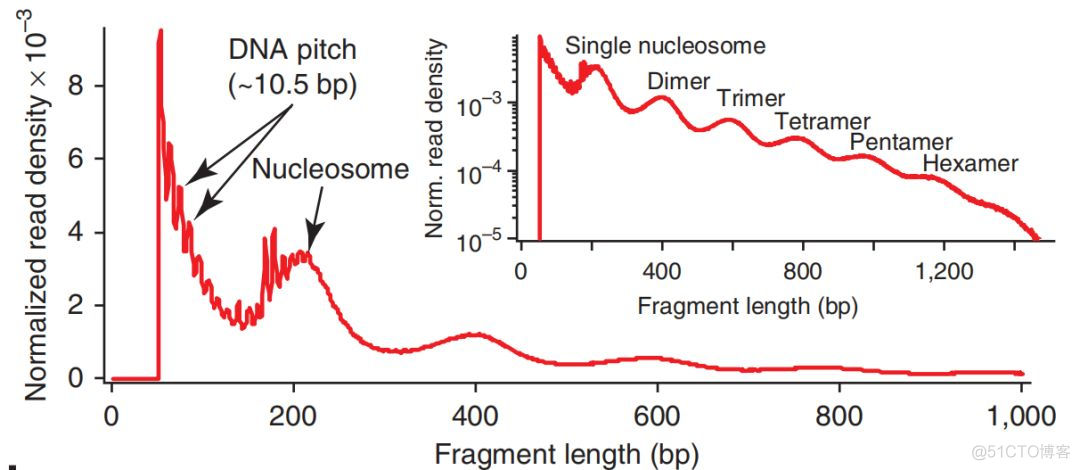
可以看到,200bp之后,插入片段的峰值有一个周期性的波动,取log之后,这个趋势更加明显。单个核小体是由146bp的DNA缠绕在组蛋白上构成的,这里的周期性表示的是不同核小体的个数,说明ATAC可以定位核小体边界。
进一步对ATAC文库的插入片段进行探究,观察不同长度的序列对应的染色质状态,结果如下
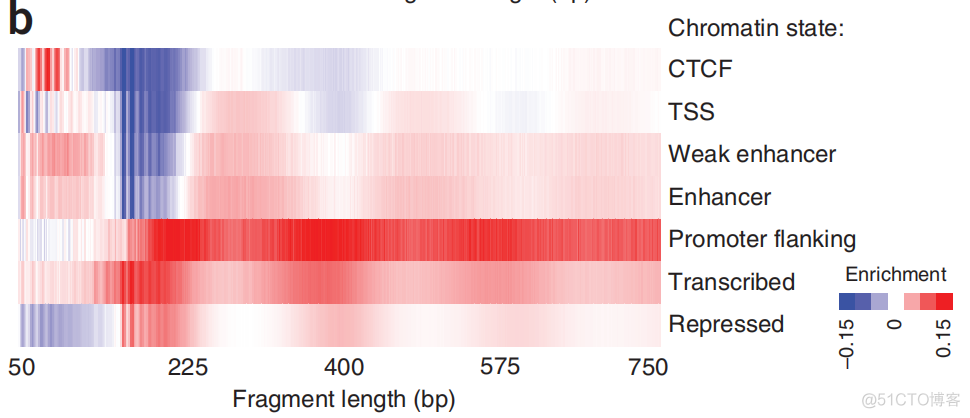
可以看到,当插入片段的长度非常短时,在CTCF结合区域富集。对于转录起始TSS区域而言,对应的插入片段长度在1到3个核小体周期的长度。而promote对应的插入片段长度则非常长。
ATAC文库中,位于两个相邻核小体之间的序列,称之为nucleosome-free fragments, 简称NRF。这部分序列的peak可以用来表征TSS的位置,如下图所示
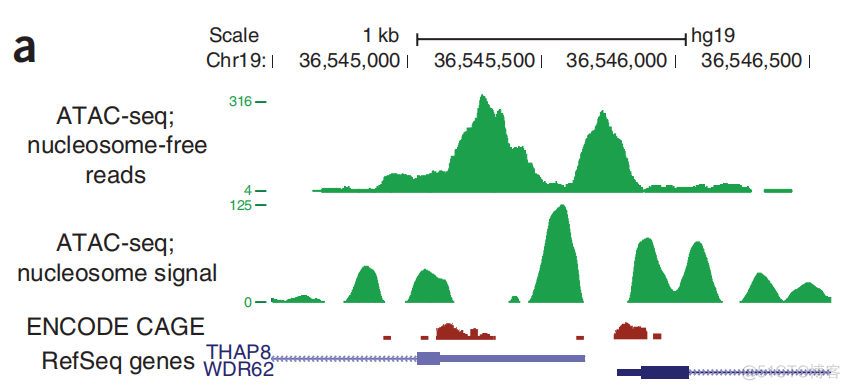
对于THAP8和WDR62两个基因而言,ENCODE CAGE测到的序列表征了这两个基因的TSS区域。可以观察到,ATAC文库中NRF序列的peak也很好的表征出了这两个TSS区域,虽然峰的中心有一定距离的偏差。
对于TSS两侧reads的分布进行统计,结果如下
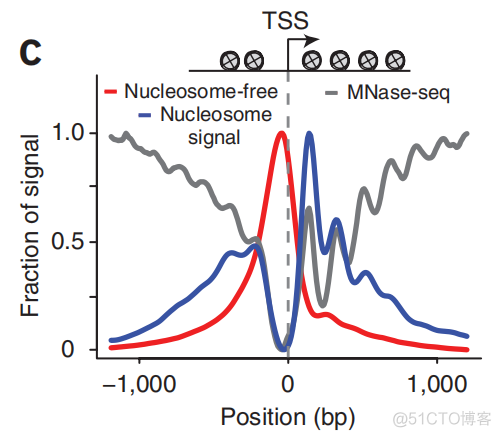
这种图主要看分布的趋势,NRF序列在TSS附近是富集的,如上图红色的峰所示。核小体边界的序列在TSS附近出也呈现了富集,但是峰值和NRF的不同。
3. ATAC揭示了转录因子结合位置与核小体的距离
利用转录因子的chip_seq数据,分析了ATAC数据中各个转录因子与核小体不同距离内序列的分布情况,结果如下
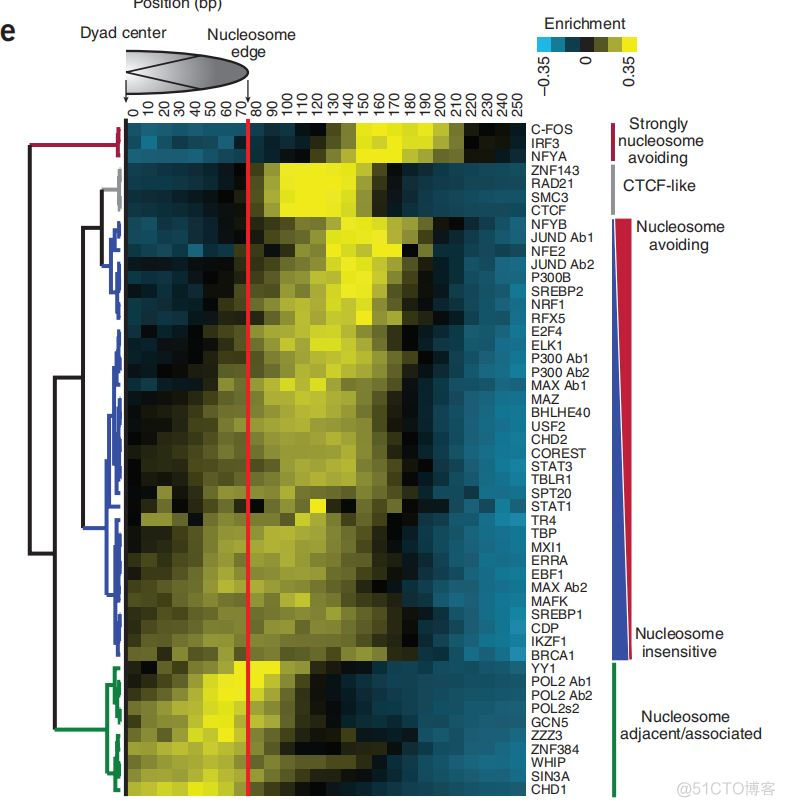
热图的每一行代表一个转录因子,通过聚类分为了4大类别,第一类距离核小体最远,为strongly nucleosome avoidting;第二类和CTCF类似,在核小体的边界附近,第四类在核小体邻近处,nucleosome adjacent/associated。
4. ATAC揭示了转录因子结合位置
chip_seq富集的是蛋白结合区域的序列,而转座酶富集的序列是蛋白两侧的序列。利用序列的分布趋势,通过ATAC也可以检测到蛋白结合区域,结果如下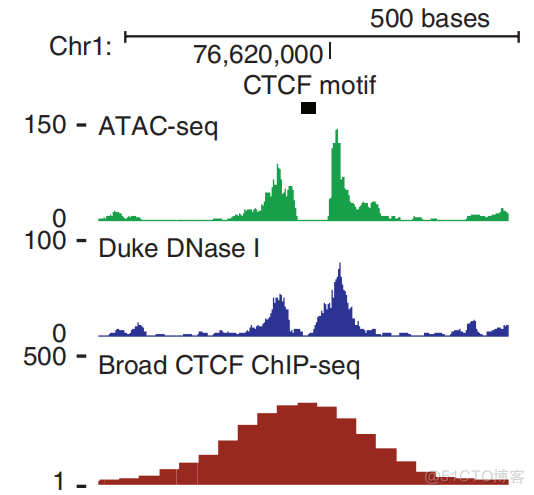
对于CTCF而言,chip_seq的峰为其结合区域,最中心的位置为对应的motif。同样的位置,在ATAC和DNase文库中,motif处没有reads,而两侧呈现富集,通过这样的趋势,也可以定位motif位置。对于某种转录因子,通过分析其motif两侧序列的分布,可以分析出文库中是否检测到了该转录因子的结合,从而识别细胞中正在发挥调控作用的转录因子。
ATAC一次获取全基因组范围内的开放染色质序列,包含的转录因子数量是非常多的。文章中通过这种方法识别到了89个转录因子,部分结果如下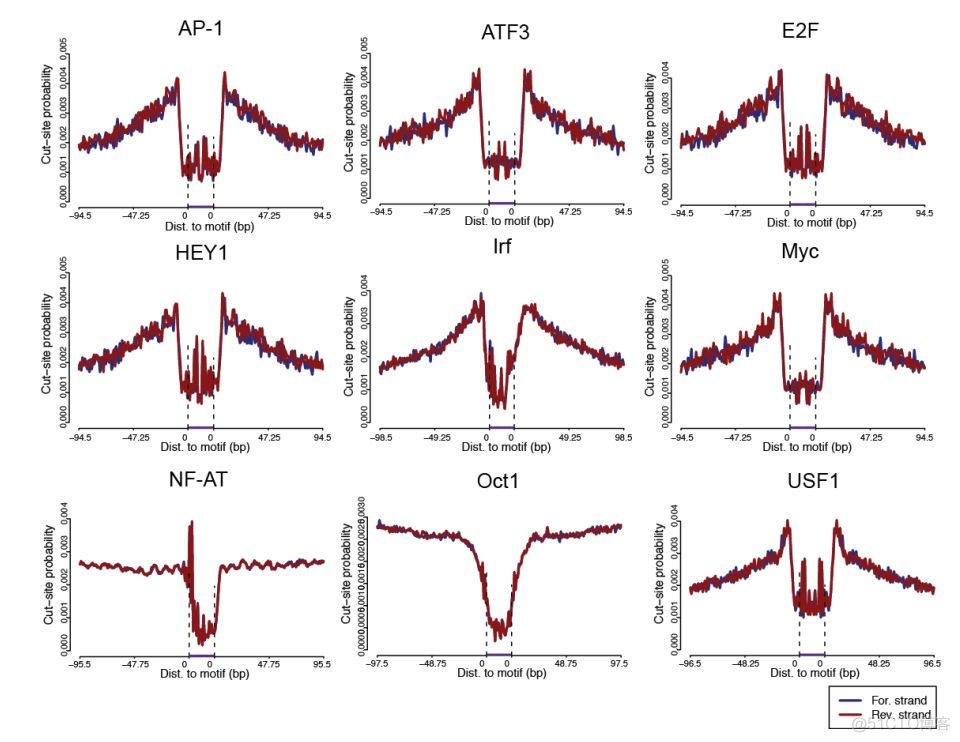
5. ATAC可以用于分析临床样本
ATAC建库周期非常短,可以实时反映样本的表观遗传信息,所以可以用于临床检测。在文章中对某个志愿者的血液样本进行了ATAC分析,首先是建库周期,统计如下
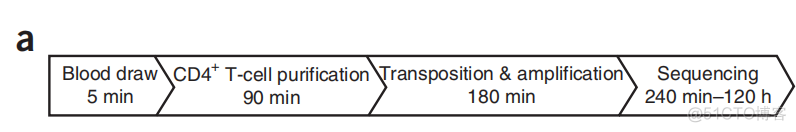
4个小时之内完成建库,然后上机测序,在上机测序的周期内,得到的表观遗传图谱是恒定的,如下所示
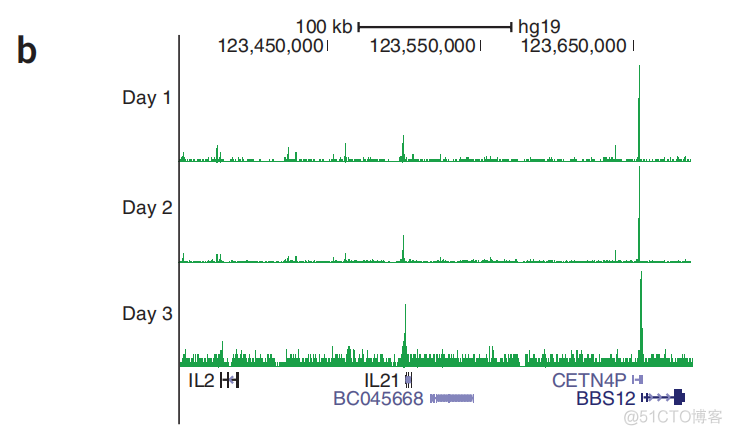
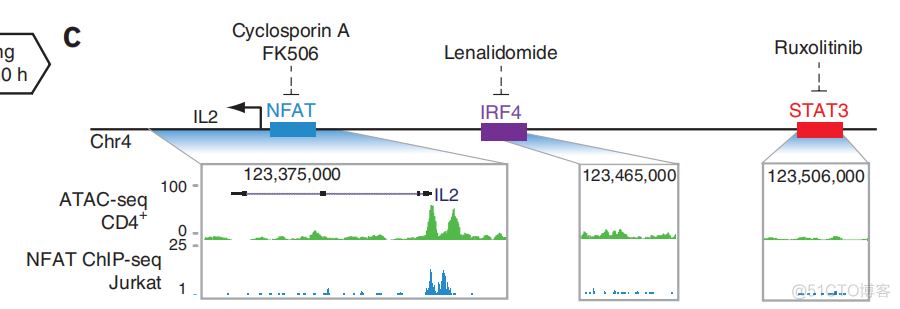
最终通过ATAC_seq, 发现了3个理论药物靶点中,只有1个靶点在患者细胞内是激活的,示意如下
ATAC在全基因组范围内捕获开放染色质区域的序列,可以识别细胞内正发挥调控功能的转录因子。由于其周期短,重复性高的特点,既可以用于基础研究,而可以处理临床样本。
·end·

生物信息入门
只差这一个
公众号