ATAC使用Tn5转座酶来完成文库的构建工作,Tn5转座酶在连接adapter序列时,会存在9bp的gap,如下图所示
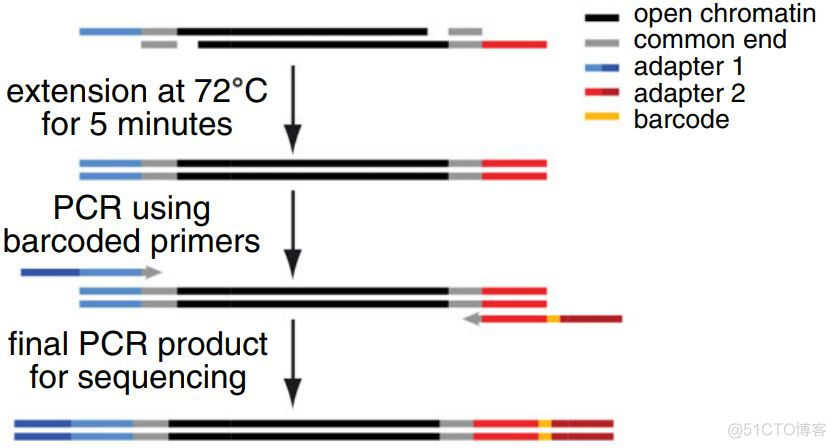
上图来自来文献ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide
从图上可以看出,为了填补gap区域,有一个5分钟的延伸过程,gap补齐之后在进行PCR扩增。
Tn5转座酶的这一特性对于ATAC的分析产生了重大影响,在ATAC中我们通过peak区域在染色体上的位置来判断蛋白结合区域,而peak区域的识别是根据序列在基因组上的比对位置得到的。
在下机数据中,序列是经过了gap补齐的,不是最初始的断裂点了。利用补齐之后的下机序列,我们得到的peak位置和真实的切割位置之间存在了偏倚,为了还原真实的染色质开放区域,在进行peak calling之前,我们需要对reads比对的参考基因组位置进行偏移。
在以下链接中,指出了偏移的具体操作
https://galaxyproject.github.io/training-material/topics/epigenetics/tutorials/atac-seq/tutorial.html
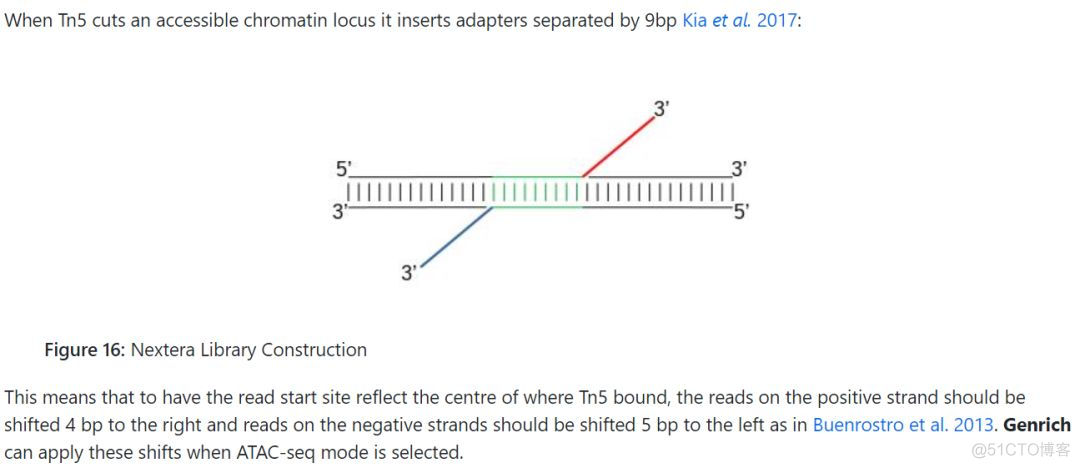
具体的,对于正链上的reads需要向右偏移4bp, 比对的起始位置加4,对于负链上的reads, 则向左偏移5bp, 比对的起始位置减5bp。在Encode给出的ATAC pipeline中,对于原始的bam文件,首先利用bedtools转换成bed文件,只保留reads比对上参考基因组的位置,然后再进行比对位置的偏移,具体的代码如下
def tn5_shift_ta(ta, out_dir):prefix = os.path.join(out_dir,
os.path.basename(strip_ext_ta(ta)))
shifted_ta = '{}.tn5.tagAlign.gz'.format(prefix)
cmd = 'zcat -f {} | '
cmd += 'awk \'BEGIN {{OFS = "\\t"}}'
cmd += '{{ if ($6 == "+") {{$2 = $2 + 4}} '
cmd += 'else if ($6 == "-") {{$3 = $3 - 5}} print $0}}\' | '
cmd += 'gzip -nc > {}'
cmd = cmd.format(
ta,
shifted_ta)
run_shell_cmd(cmd) return shifted_ta
对于chip_seq的数据分析,拿到bam文件之后直接peak calling就可以了,对于ATAC_seq而言,一定要偏移之后再进行peak calling。
·end·

一个只分享干货的
生信公众号