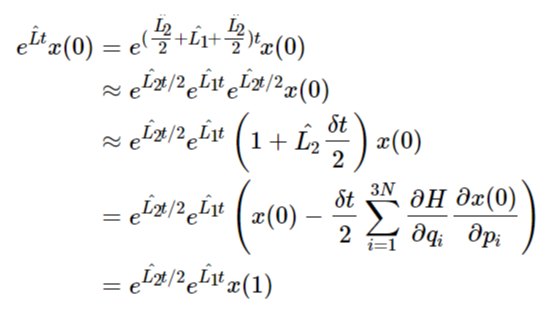 本文延续历史上分子动力学模拟演化算法的发展顺序,分别讲述了Verlet、LeapFrog和Velocity-Verlet三个算法的形式,并且结合刘维尔方程,推导了Velocity-Verlet算法中的三个演化步骤的内在含义。三种不同的演化算法,都有不同的初始依赖,而对于计算过程而言,越多的初始依赖,就会涉及到越多的参数存储问题。一个好的演化算法,只需要依赖于少量的参数,而具备较高的精度。
技术背景
本文延续历史上分子动力学模拟演化算法的发展顺序,分别讲述了Verlet、LeapFrog和Velocity-Verlet三个算法的形式,并且结合刘维尔方程,推导了Velocity-Verlet算法中的三个演化步骤的内在含义。三种不同的演化算法,都有不同的初始依赖,而对于计算过程而言,越多的初始依赖,就会涉及到越多的参数存储问题。一个好的演化算法,只需要依赖于少量的参数,而具备较高的精度。
技术背景
我们说分子动力学模拟是一个牛顿力学的过程,在使用量子化学的手段或者深度学习的方法或者传统的力场方法,去得到某个时刻某个位置的受力之后,就可以获取下一步的整个系统的状态信息。这个演化的过程所使用的算法,也在历史上演化了多次,从1967年的Verlet算法,到后来的Leap-Frog算法,再到Velocity-Verlet算法。我们可以先看一看这三种算法的形式,再从刘维尔方程出发,看看Velocity-Verlet算法的由来。
Verlet算法我们把\(r(t+\delta t)\)和\(r(t-\delta t)\)看做是两个函数,分别对\(r(t+\delta t)\)和\(r(t-\delta t)\)在时刻\(t\)处进行二阶泰勒展开有:
\[r(t+\delta t)=r(t)+v(t)\delta t+\frac{F(t)}{2m}\delta t^2\\ r(t-\delta t)=r(t)-v(t)\delta t+\frac{F(t)}{2m}\delta t^2 \]将上面第二个公式中的\(v(t)\delta t\)项移到左侧,把\(r(t-\delta t)\)移到右侧,再代入到第一个公式中,就可以得到下一步的坐标:
\[r(t+\delta t)=2r(t)-r(t-\delta t)+\frac{F(t)}{m}\delta t^2+O(\delta t^4) \]然后再把1、2两个公式相减,就可以得到当前时刻的速度:
\[v(t)=\frac{r(t+\delta t)-r(t-\delta t)}{2\delta t}+O(\delta t^2) \]到这里就得到了Verlet算法的更新步骤,过程也非常的简单。但是有个比较致命的问题是,Verlet算法的更新中,不仅仅需要到上一步的坐标位置,还需要用到上上一步的坐标位置,这就有可能导致两个问题:
- 第一步的更新,没有上上一步的信息;
- 在算法执行的过程中,每次迭代不仅仅要存储上一步的坐标位置,还需要额外存储上上一步的位置,更新较为麻烦,也会占据额外的空间。
目前这种传统的Verlet算法应用已经较少,主要还是使用接下来要讲到的Leap-Frog算法和Velocity-Verlet算法。
Leap-Frog算法在蛙跳法中,引入了另外一个概念:用两点之间的中间时刻的速度近似为两个点之间的平均速度,这样就可以得到一个坐标更新公式:
\[r(t+\delta t)=r(t)+v(t+\frac{\delta}{2})\delta t \]其中半步的速度是基于上一个半步的速度来更新的:
\[v(t+\frac{\delta t}{2})=v(t-\frac{\delta t}{2})+\frac{F(t)\delta t}{m}=v(t-\frac{\delta t}{2})-\frac{\partial V}{\partial r(t)}\cdot\frac{\delta t}{m} \]在上面的方程中已经用势能对坐标的偏导来替代力的计算,这也跟哈密顿力学中只有势能项显含坐标有关。虽然到这里我们已经完成了坐标和速度的更新,但是速度和坐标之间是不同步的,为此我们还需要用两个半步速度取平均来计算一个时间同步的速度:
\[v(t+\delta t)=\frac{v(t+\frac{\delta t}{2})-v(t-\frac{\delta t}{2})}{2} \]由于这里只涉及到前半步的速度,而不涉及到前一步的坐标,因此Leap-Frog算法在实际应用场景中有着较为广泛的使用。
刘维尔方程与Velocity Verlet首先我们看一下刘维尔方程的形式:
\[\frac{d\rho(p,q,t)}{dt}=\frac{\partial\rho}{\partial t}+\hat{L}\rho=0 \]其中刘维尔算子\(\hat{L}\)的形式为:
\[\hat{L}=\hat{L_1}+\hat{L_2}=\sum_{i=1}^{3N}\left(\frac{\partial H}{\partial p_i}\frac{\partial}{\partial q_i}-\frac{\partial H}{\partial q_i}\frac{\partial}{\partial p_i}\right) \]其中写成\(\hat{L_1}\)和\(\hat{L_2}\)的形式也是为了方便后面做Trotter分解:
\[\hat{L_1}=\sum_{i=1}^{3N}\frac{\partial H}{\partial p_i}\frac{\partial}{\partial q_i}\\ \hat{L_2}=-\sum_{i=1}^{3N}\frac{\partial H}{\partial q_i}\frac{\partial}{\partial p_i} \]写完刘维尔方程的表述之后,我们再回过头看看刘维尔方程的物理含义,这里的密度函数\(\rho(p,q,t)\)是指在相空间中粒子的分布密度,对于整体的积分有:
\[N=\int\rho(p,q,t)dqdp \]这里的\(N\)所表示的就是整个系统的总粒子数。因此,实际上刘维尔方程所表述的内容就是:分布函数沿着相空间的任何轨迹是常数。
Trotter-Suzuki分解我们首先需要回顾一个知识点,虽然对于两个常数\(a,b\)来说,其加和的指数可以等于指数的乘积,即\(e^{a+b}=e^{a}e^{b}\),但如果是对于两个矩阵\(A,B\)的话,类似的等式往往是不成立的。而Trotter-Suzuki公式,将其表示为一个显式的误差:
\[e^{\sum_{j=1}^{m}H_jt}=\prod_{j=1}^{m}e^{H_jt}+O(m^2t^2) \]此时我们再回顾刘维尔算子的分解:\(\hat{L}=\hat{L_1}+\hat{L_2}\),再进一步将其分解为:\(\hat{L}=\frac{\hat{L_2}}{2}+\hat{L_1}+\frac{\hat{L_2}}{2}\),至于为什么用这个形式来分解,我也不懂,也许是尝试出来的。基于这个形式的分解,我们将其代入到刘维尔算子的演化中。定义一个广义参量\(x(t)=\{p(t),q(t)\}\),则刘维尔算子对该参量的演化为:
\[\begin{align} e^{\hat{L}t}x(0)&=e^{(\frac{\hat{L_2}}{2}+\hat{L_1}+\frac{\hat{L_2}}{2})t}x(0)\\ &\approx e^{\hat{L_2}t/2}e^{\hat{L_1}t}e^{\hat{L_2}t/2}x(0)\\ &\approx e^{\hat{L_2}t/2}e^{\hat{L_1}t}\left(1+\hat{L_2}\frac{\delta t}{2}\right)x(0)\\ &=e^{\hat{L_2}t/2}e^{\hat{L_1}t}\left(x(0)-\frac{\delta t}{2}\sum_{i=1}^{3N}\frac{\partial H}{\partial q_i}\frac{\partial x(0)}{\partial p_i}\right)\\ &=e^{\hat{L_2}t/2}e^{\hat{L_1}t}x(1) \end{align} \]观察上述推导过程的倒数第二步,因为\(x(t)=\{p(t),q(t)\}\),并且在相空间中所有的\(p_i\)是正交的关系,因此\(\frac{\partial x(0)}{\partial p_i}\)得到的结果全为1。又因为在哈密顿力学中有\(-\frac{\partial H}{\partial q}=\frac{dp}{dt},\frac{\partial H}{\partial p}=\frac{dq}{dt}\)。因此,假定\(x(0)=\{p_i(t_0),q_i(t_0)\},i=1,2,...,3N\),则\(x(1)=\{p_i(t_0)+\frac{dp(t_0)}{dt}\frac{\delta t}{2},q_i(t_0)\},i=1,2,...,3N\)。使用类似的方法,我们继续往下推导:
\[\begin{align} e^{\hat{L}t}x(0)&\approx e^{\hat{L_2}t/2}e^{\hat{L_1}t}x(1)\\ &=e^{\hat{L_2}t/2}\left(x(1)+\delta t\sum_{i=1}^{3N}\frac{\partial H}{\partial p_i}\frac{\partial x(1)}{\partial q_i}\right)\\ &=e^{\hat{L_2}t/2}x(2) \end{align} \]其中\(x(2)=\{p_i(t_0)+\frac{dp(t_0)}{dt}\frac{\delta t}{2},q_i(t_0)+\frac{dq(t_0)}{dt}\delta t\},i=1,2,...,3N\),同样的方法,再完成最后一步的分解:
\[\begin{align} e^{\hat{L}t}x(0)&\approx e^{\hat{L_2}t/2}x(2)\\ &=x(2)-\frac{\delta t}{2}\sum_{i=1}^{3N}\frac{\partial H}{\partial q_i}\frac{\partial x(2)}{\partial p_i}\\ &=x(3) \end{align} \]需要注意的是,虽然在前面\(x(0)\rightarrow x(1)\)的演化中共轭动量项在\(\hat{L_2}\)的作用下发生了变化,但是显含的动量项保持不变,因此这里的偏导项得到的结果依然是1,那么就有\(x(3)=\{p_i(t_0)+\frac{dp(t_0)}{dt}\delta t,q_i(t_0)+\frac{dq(t_0)}{dt}\delta t\},i=1,2,...,3N\)。到这一步,问题逐渐露出端倪,我们发现在刘维尔算子的作用下,经过Trotter-Suzuki分解和Taylor展开的操作,正则坐标\(q\)和共轭动量\(p\)已经完成了一个时间单位\(\delta t\)的演化,正对应到分子动力学模拟中的单步演化。
Velocity Verlet算法参考上一个章节中刘维尔算子的演化过程\(x(0)\rightarrow x(1)\),我们可以先将速度进行半步演化:
\[v(t+\frac{\delta t}{2})=v(t)+\frac{F(t)}{m}\frac{\delta t}{2}+O(\delta t^3) \]参考\(x(1)\rightarrow x(2)\)过程,我们可以将坐标进行单步演化:
\[r(t+\delta t)=r(t)+v(t+\frac{\delta t}{2})\delta t+O(\delta t^4) \]最后参考\(x(2)\rightarrow x(3)\)的过程,将半步演化的速度再同步到单步演化:
\[v(t+\delta t)=v(t+\frac{\delta t}{2})+\frac{F(t+\delta t)}{m}\frac{\delta t}{2}+O(\delta t^2) \]这个过程最漂亮的地方在于,演化的参数不依赖于上一步或者上半步的任何参数,只需要具备了\(v(t),r(t)\)即可演化得到\(v(t+\delta t),r(r+\delta t)\),当然,这里面需要用量子化学或者深度学习或者是力场参数的形式,去分别计算得\(t\)和\(t+\delta t\)时刻的作用力。
总结概要本文延续历史上分子动力学模拟演化算法的发展顺序,分别讲述了Verlet、LeapFrog和Velocity-Verlet三个算法的形式,并且结合刘维尔方程,推导了Velocity-Verlet算法中的三个演化步骤的内在含义。三种不同的演化算法,都有不同的初始依赖,而对于计算过程而言,越多的初始依赖,就会涉及到越多的参数存储问题。一个好的演化算法,只需要依赖于少量的参数,而具备较高的精度。
版权声明本文首发链接为:https://www.cnblogs.com/dechinphy/p/liouville.html
作者ID:DechinPhy
更多原著文章请参考:https://www.cnblogs.com/dechinphy/
打赏专用链接:https://www.cnblogs.com/dechinphy/gallery/image/379634.html
腾讯云专栏同步:https://cloud.tencent.com/developer/column/91958
“留一手”加剧内卷,“讲不清”浪费时间。