FUSION是一款进行TWAS分析的软件,对应的文章发表在nature genetic上,链接如下
https://www.nature.com/articles/ng.3506
软件的官网如下
http://gusevlab.org/projects/fusion/
该软件同时支持对单个样本和多个样本的gwas cohort中的样本进行基因型填充,对应的流程图如下
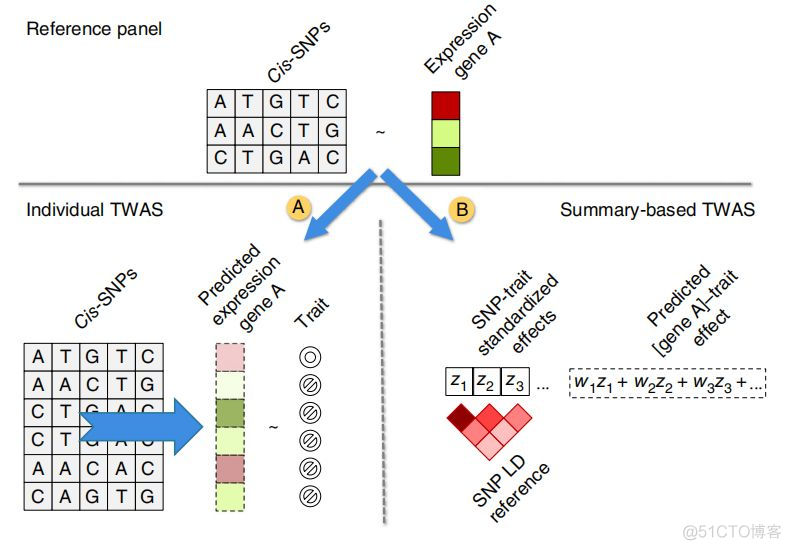
首先对reference panel中进行建模,对于每个基因,筛选对应的cis-snp, 即在该基因上下游1Mb范围内的snp位点,通过不同的模型构建cis-SNPs和对应的gene之间的关系,接下来对gwas cohort中的样本进行填充,如上图中A所示,对于单个样本,根据其cis-SNPs的基因分析结果,预测对应基因的表达量,然后与表型性状进行关联分析;如上图中B所示,对于大量样本的gwas summary数据,考虑了SNP的连锁情况,根据cis-SNP和gene的关系,直接预测基因和性状之间的关联效应。
该软件采用R语言进行开发,基本用法如下
1. 利用reference panel建模
通过不同的模型,计算cis-SNPs和基因的关系,代码如下
Rscript FUSION.compute_weights.R \--bfile $INP \
--tmp $TMP \
--out $OUT \
--models top1,blup,bslmm,lasso,enet
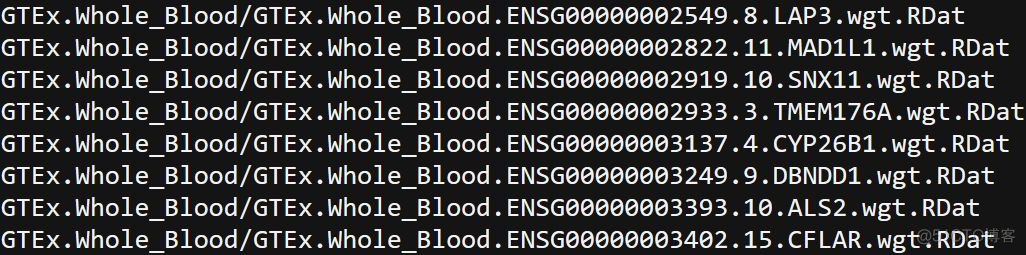
bfile参数指定plink格式的基因分型结果,tmp参数指定临时文件存放的目录,out参数指定输出文件的目录,运行成功后,在输出结果的目录,每个基因都会生成1个后缀为RDat的文件。接下来需要生成一个profile文件,首先准备一个list文件,每一行为一个基因对应的RData文件的路径,示意如下
运行以下命令生成profile
Rscript utils/FUSION.profile_wgt.R GTEx.Whole_Blood.list对于GTEx, TCGA等公共数据,官网提供了事先构建好的模型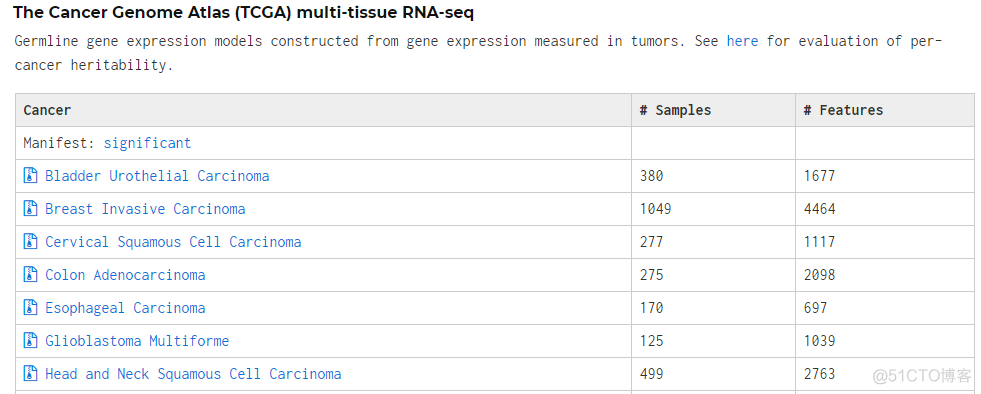
根据自己的细胞、组织类型,下载对应的数据集。
2. 对gwas cohort进行TWAS分析
代码如下
Rscript FUSION.assoc_test.R \--sumstats PGC2.SCZ.sumstats \
--weights ./WEIGHTS/GTEx.Whole_Blood.pos \
--weights_dir ./WEIGHTS/ \
--ref_ld_chr ./LDREF/1000G.EUR. \
--chr 22 \
--out PGC2.SCZ.22.dat
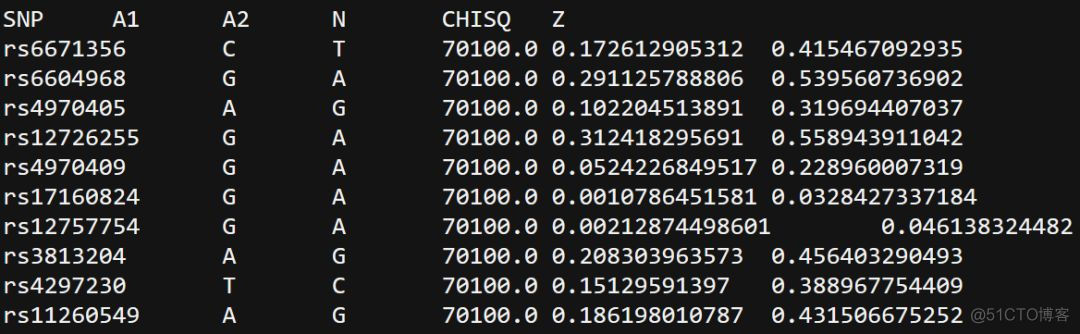
sumstats参数指定gwas队列样本的SNP分型结果,文件内容示意如下
weights指定基因的位置信息,内容示意如下
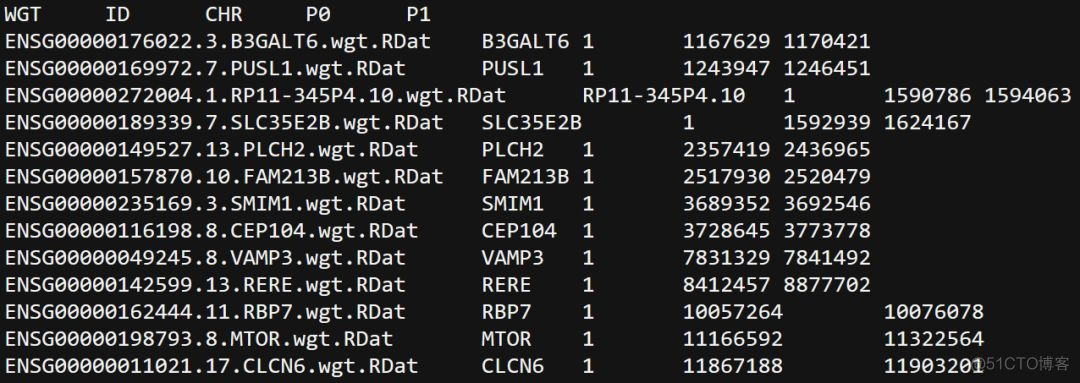
weights_dir参数指定基因RDat文件所在的目录,ref_ld_chr指定LD文件, 通常使用1000G的LD reference data, chr参数指定分析的染色体,out参数指定输出的结果。
输出结果列数很多,官网给出每一列的详细解释
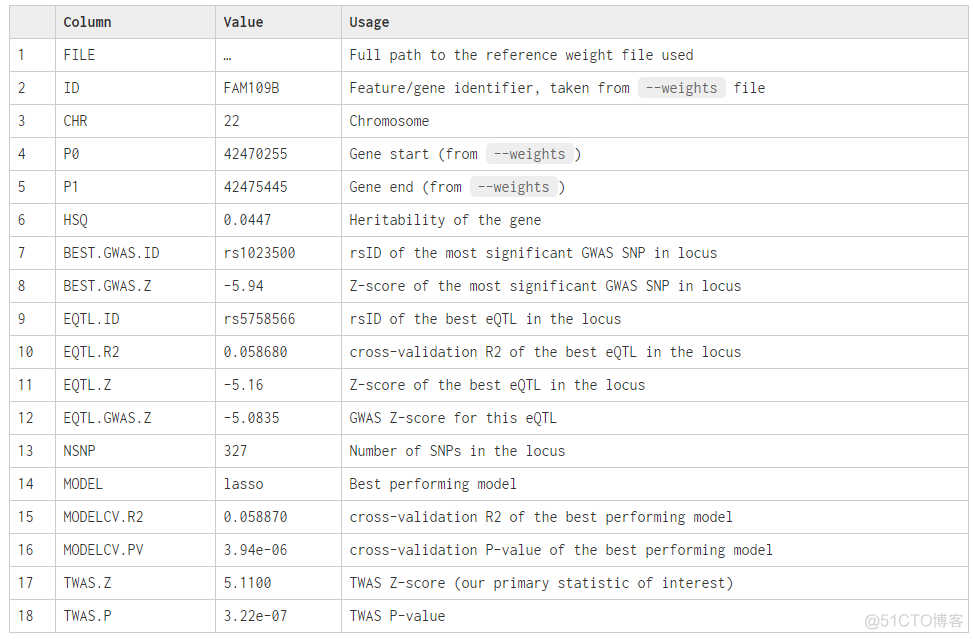
FUSION使用的较为广泛,还有人用这个软件分析了很多gwas summary数据,做成了数据库twas-hub, 后续在详细介绍这个数据库。
·end·

生物信息入门
只差这一个
公众号