欢迎关注”生信修炼手册”!
GCTA全称如下
Genome-wide Complex Trait Analysis
是一款针对复杂性状的SNP分型数据进行分析的软件,开发的初衷是用于评估表型变异中遗传变异所占的比例,即评估遗传力的大小, 后来逐渐扩展到更多的功能,官网如下
https://cnsgenomics.com/software/gcta
本文主要介绍使用该软件分析样本间亲缘关系的方法。GWAS分析要求样本间是相互独立的,所以在质控阶段,会根据样本间的亲缘关系,剔除亲缘关系较近的样本。样本的亲缘关系有多种定义和求解方式, 其中通过plink计算IBD距离是最经典的一种,而在GCTA中,则采用了另外一套思路。
GCTA中定义的样本亲缘关系计算公式如下
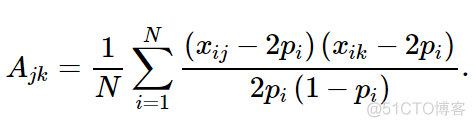
A表示样本j和k之间的亲缘关系,N表示SNP位点总数,i表示某个SNP位点,xij表示样本j中SNP位点i的分型结果,用0,1,2表示,代表ref allele的个数, 假设位点突变情况为A突变到G, 则AA对应2,AG对应1,GG对应0,xik同理,表示样本k中SNP位点i的分型结果;pi表示ref allele在所有样本中的频率。
不同的取值表示不同的亲缘关系
- 1 for MZ twins / duplicated samples
- 0.5 for 1st degree relatives (e.g. full-sibs or parent-offspring)
- 0.25 for 2nd degree relatives (e.g. grandparent-grandchild)
- 0.125 for 3rd degree relatives (e.g. cousins)
对于两两样本间的亲缘关系,可以用一个矩阵表示,即genetic relationship matrix, 简称GRM。使用GCTA计算GRM的代码如下
gcta64 --bfile test --autosome --make-grm --out test输入数据为plink二进制格式的文件,--autosome表示只使用常染色体上的SNP位点,--make-grm表示计算GRM矩阵,--out指定输出结果前缀。该方法输出的结果为二进制格式,更适合用于下游分析。如果想查看样本间对应的亲缘关系,可以用以下方式
gcta64 --bfile test --autosome --make-grm-gz --out test此时样本间亲缘关系会输出在一个压缩文件中,其内容示意如下
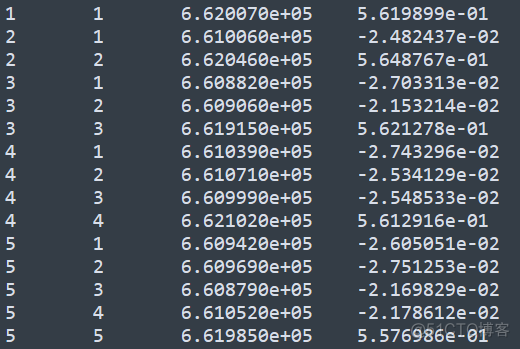
前两列表示样本的编号,第三列表示两个样本中都分型成功的SNP位点个数,第四列表示两个样本间的亲缘关系值。其排列方式示意如下
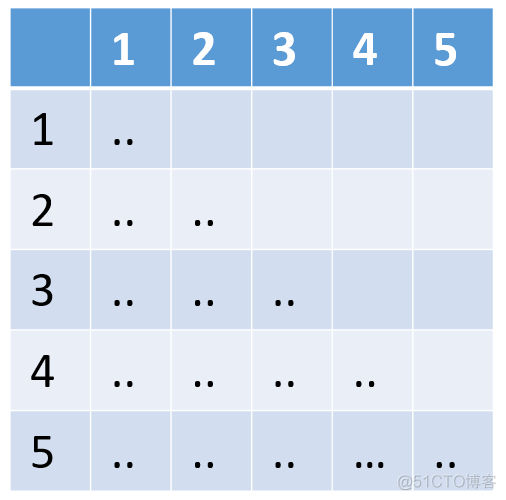
GRM矩阵是一个方阵,是对称矩阵,所以用下三角矩阵表示。对应到上述文件中,从第一行开始记录对应的值。样本编号对应的样本名可以在后缀为id的文件中找到,其内容示意如下
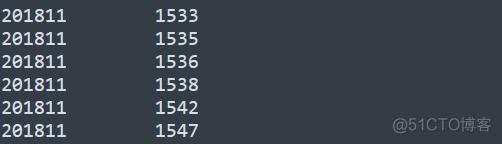
第一列为family id, 第二列为individual Id, 样本所在的行数就是对应的编号。计算出样本间的亲缘关系之后,我们可以绘制如下所示的密度分布图,来查看其分布
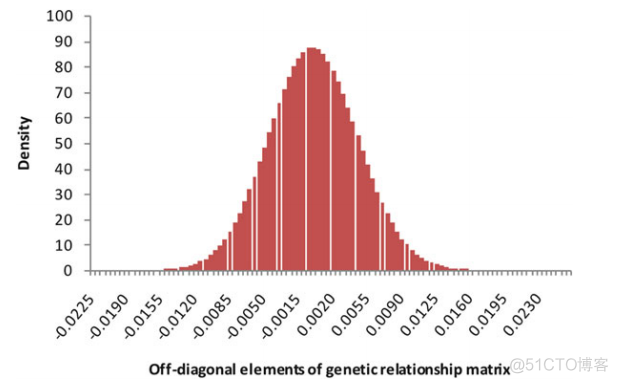
针对亲缘关系大的情况, 我们进行过滤,比如设定阈值为0.125, 亲缘关系大于该阈值的样本间就需要剔除其中一个样本。GCTA采用迭代的方式进行剔除,保证剩余样本的个数最大化。代码如下
gcta64 --grm test --grm-cutoff 0.025 --make-grm --out test_rm025可以看到GRM矩阵是GCTA数据分析的核心,后续的分析都需要依赖这个矩阵,所以删除样本也是在针对GRM矩阵进行操作,删除对应样本后,生成一个新的矩阵。
质控之后,就可以进行下游分析了,具体的用法在后续文章中再详细介绍。
·end·

生物信息入门
只差这一个
公众号