MACS2作为使用最广泛的peak calling软件,在v2版本中添加了差异peak分析的功能,所有的子命令功能描述如下
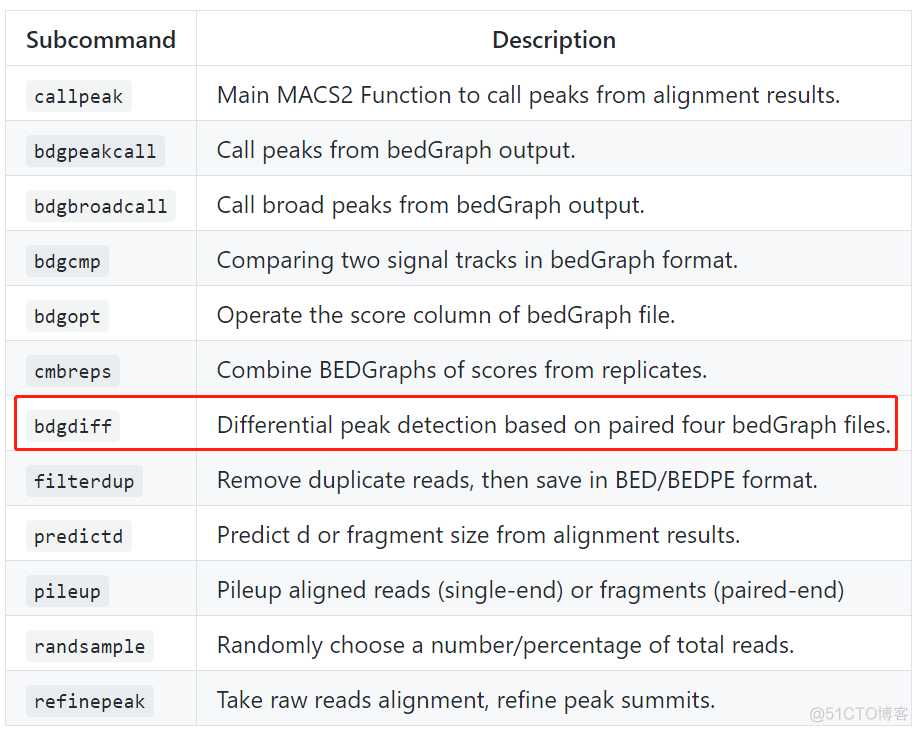
通过bdgdiff子命令来进行差异peak分析, 该命令不需要基于已有的peak calling结果,只需要输入每个样本对应的bedGraph格式的文件。需要注意的是,该命令只针对两个样本间的差异peak进行设计,适用于没有生物学重复的情况。
对于使用macs2来进行差异peak的完整流程,官方给出了详细的说明文档,链接如下
https://github.com/taoliu/MACS/wiki/Call-differential-binding-events
可以分为以下3步
1. 预测插入片段长度
通过predictd子命令可以预测样本的fragment size,命令如下
macs2 predictd -i input.bam2. peak calling
在peak calling时,需要添加-B参数,这样才可以输出样本对应的bedgraph文件,同时需要保证peak calling时采用一致的--extsize的值,就是第一步预测出来的数值,取多个样本的均值即可。官方也给出了推荐值,对于大多数的转录因子chip_seq数据,推荐值为200, 对于大部分组蛋白修饰的chip_seq数据,推荐值为147,命令如下
# condition1macs2 callpeak -B -t cond1_ChIP.bam -c cond1_Control.bam -n cond1 --nomodel --extsize 120
# condition2
macs2 callpeak -B -t cond1_ChIP.bam -c cond1_Control.bam -n cond1 --nomodel --extsize 120
在运行这一步的时候,会输出每个样本过滤之后的reads数目,示意如下
# tags after filtering in treatment: 19291269# tags after filtering in control: 12914669
这个数值在差异分析中会用到,所以要记录下来。
3. 差异peak分析
命令如下
macs2 bdgdiff --t1 cond1_treat_pileup.bdg --c1 cond1_control_lambda.bdg --t2 cond2_treat_pileup.bdg \--c2 cond2_control_lambda.bdg --d1 12914669 --d2 14444786 -g 60 -l 120 --o-prefix diff_c1_vs_c2
其中-d1和-d2的值就是第二步运行时输出的reads数目,-o参数指定输出文件的前缀。运行成功后,会产生3个文件
其中, con1.bed保存了在condition1中上调的peak, con2.bed保存了在condition2中上调的peak, common.bed文件中保存的是没有达到阈值的,非显著差异peak。
上述3个文件格式是完全相同的,最后一列的内容为log10 likehood ratio值,用来衡量两个条件之间的差异,默认阈值为3,大于阈值的peak为组间差异显著的peak, 这个阈值可以通过-c参数进行调整。
·end·

一个只分享干货的
生信公众号