对于ATAC_seq, chip_seq等抗体富集型文库而言,peak calling是分析的第一步。通过peak calling,可以得到抗体富集的区域,这些区域有对应的生物学功能,在chip_seq中,可以是转录因子结合区或者发生组蛋白修饰的区域,ATAC中对应的就是开放染色质区域。
上述各种测序研究的对象都是基因的表达调控,peak calling帮助我们得到调控片段的染色体位置,可以构建潜在的调控网络。众所周知,基因表达调控是一个动态变化的过程,不同生长阶段,不同实验条件的样本间,其调控元件是会存在差异的,而研究这个动态过程,其意义更加重大,对应到数据上,peak caling之后,我们要做的就是peak 的差异分析。
peak差异分析的工具很多,不同软件的结果不尽相同,如何选择是一个难题。在下列文章中,以chip_seq数据为例,针对已经发表的多个peak差异分析工具进行了探索

文章链接如下
http://bioinfo.sibs.ac.cn/shaolab/pdf/2017%20quantitative%20biology;%20review%20of%20chipseq%20differential%20binding%20analysis.pdf
peak 差异分析与peak caling的结果紧密相依,在上述文献中,将peak差异分析总结为了两大类,示意如下
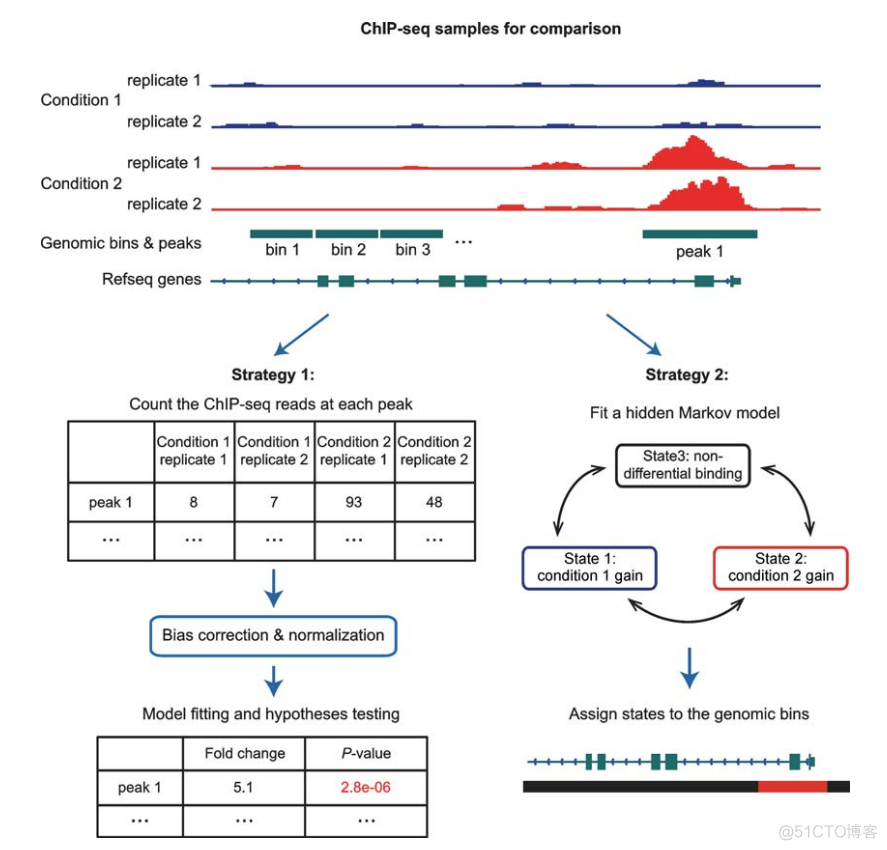
第一类,类似转录组差异分析的策略,首先基于peak calling的结果,统计peak区域在各个样本中的表达量,然后进行归一化,差异分析;第二类,采用了隐马可夫模型,将基因组的区域分为了非差异,上调,下调3种不同状态,构建3种状态间的转移矩阵,二者最大的区别就在于第二类的软件不需要依赖已有的peak calling结果。
在实验设计中,还需要考虑到一个因素就是生物学重复,虽然大多数实验都是有生物学重复的,但是没有生物学重复的情况也不可避免,这在选择对应的分析软件时要充分考虑。为了方便选取,文献中整理了如下所示的决策树
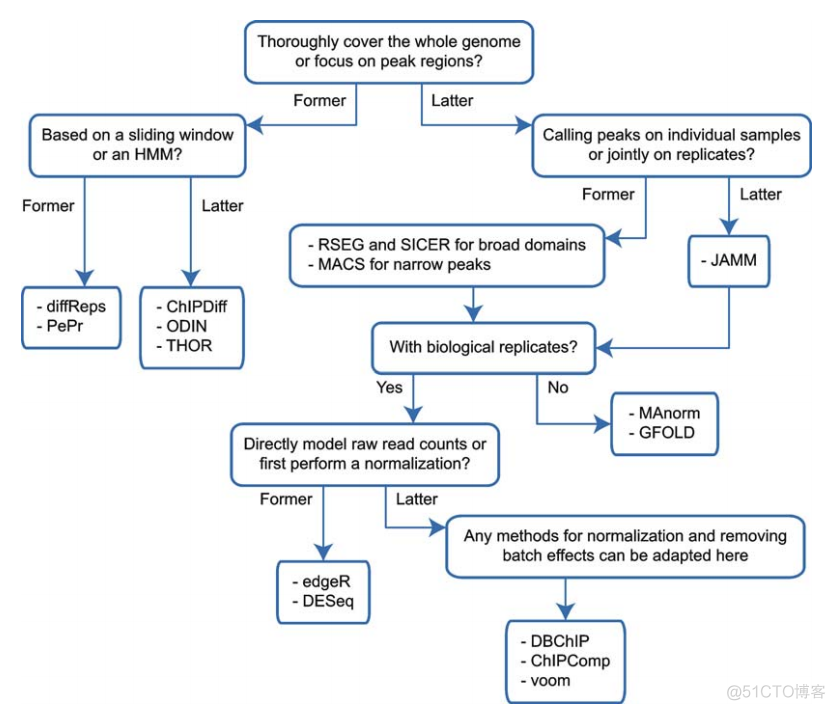
首先明确是是否基于已有的peak区域进行分析,如果不基于已有的peak区域,可以选择滑动窗口或者隐马可夫模型, 其中基于滑动窗口的软件如下
基于隐马可夫模型的软件如下
如果基于已有的peak结果进行分析,则需要根据有无生物学重复进行判断,如果没有生物学重复,可以选择MAnorm或者GFOLD软件,如果有生物学重复,而且统计的是raw count格式的表达量,则可以用转录组中常用的edgeR, DESeq进行差异分析,如果不是raw count, 则可以用DBChip, ChIPComp, voom来进行分析。
在文章中,说明了用edgeR和DESeq进行peak 差异分析的理由,将peak看做是RNA_seq中的基因,则其定量方式和差异分析可以通用。另外,文章中还提到一种很有启发的差异分析思路,用macs和scier等peak caling软件进行差异分析,这些软件的常规用法是比较实验样本和input样本的差异,进行peak calling, 在用其进行差异分析时,将对照组的样本看做input, 将实验组的样本设置为case, 然后进行peak calling,最终鉴定到的区域,可以看做是两组间的差异peak 区域。
这篇文章发表的比较早,一些些的差异peak分析软件没有包括进来,比如DiffBind, macs2的差异peak 分析功能,后续在详细介绍各个软件的用法。
·end·

一个只分享干货的
生信公众号