在数据分析中,软件经常会要求参考基因组对应的chrom.sizes文件,该文件保存了基因组中的染色体名称已经对应的长度,内容示意如下 第一列为染色体名称,第二列为染色体的长度。
在数据分析中,软件经常会要求参考基因组对应的chrom.sizes文件,该文件保存了基因组中的染色体名称已经对应的长度,内容示意如下
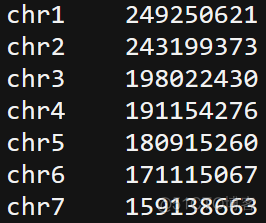
第一列为染色体名称,第二列为染色体的长度。本文介绍一下生成该文件的3种方式
1. 从UCSC下载
这种方法适用于UCSC数据库中已有的物种。以hg19为例,在UCSC的FTP中直接提供了对应的chr.sizes文件,链接如下
http://hgdownload.soe.ucsc.edu/goldenPath/hg19/bigZips/
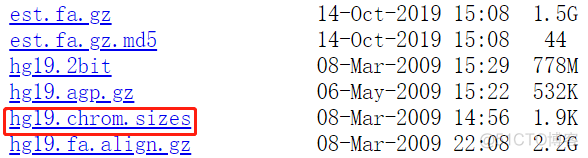
下载红框标记的chrom.sizes文件即可。
2. 利用samtools进行提取
samtools的faidx命令可以获取fasta文件中的序列长度信息,从其生成的后缀为fai的文件中可以获得chrom.sizes文件,用法如下
samtools faidx hg19.facut -f1,2 hg19.fa.fai > hg19.chrom.sizes
3. 自己写脚本进行统计
自己写脚本就比较灵活多变了,无论采用什么编程语言都可以,这里用perl给一个示例,代码如下
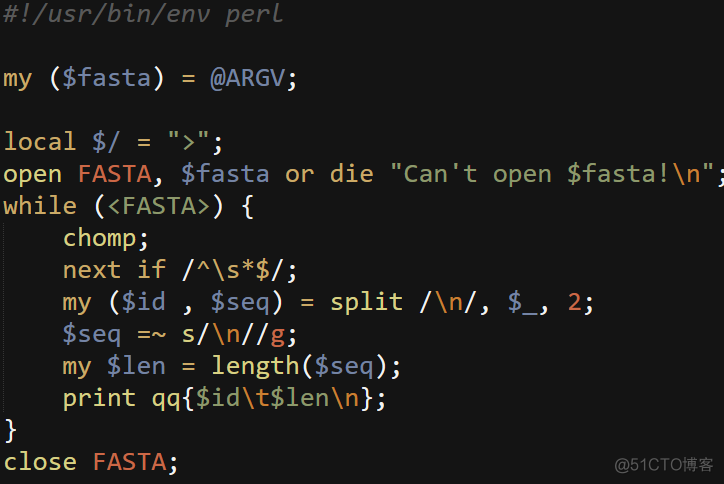
运行该脚本即可,命令如下
perl cal_chrom_sizes.pl hg19.fa > hg19.chrom.sizes第一种方法受到了UCSC数据库的限制,第二种方法运行速度块,通用性强,更加推荐使用。自己写脚本的话,就是更加的灵活,可以根据自己的需求灵活定制。
·end·

一个只分享干货的
生信公众号