ENCODE称之为基因组百科全书,该数据库包含了基因组学,转录组学,表观遗传学等许多组学的数据。在提供公共数据的同时,还开源了许多组学数据分析的pipeline,当然也包含了ATA
ENCODE称之为基因组百科全书,该数据库包含了基因组学,转录组学,表观遗传学等许多组学的数据。在提供公共数据的同时,还开源了许多组学数据分析的pipeline,当然也包含了ATAC数据分析的pipeline, 对应的网址如下
https://www.encodeproject.org/atac-seq/
目前最新版的pipeline网址如下
https://github.com/ENCODE-DCC/atac-seq-pipeline
ENCODE不仅给出了pipeline, 同时还根据处理ATAC数据的经验,给出了质控的标准,非常值得参考。质控标准有以下几点
实验设计时需要考虑生物学重复,每组至少2个生物学重复,对于实现材料有限,无法达到2个生物学重复的样本,也要设计至少2个技术重复每个样本需要至少25M的有效序列,这里的有效序列指的是去除PCR重复,去除线粒体之后的序列,单位是fragments。对于单端测序而言,需要至少25M的reads, 对于双端测序而言,需要至少50M的readsmapping比例,即比对上参考基因组的reads所占比例大于95%, 当然80%以上也可以接受文库复杂度, 和chip_seq类似,采用PBC1, PBC2, NRF共3个指标来描述文库复杂度,对应的标准如下
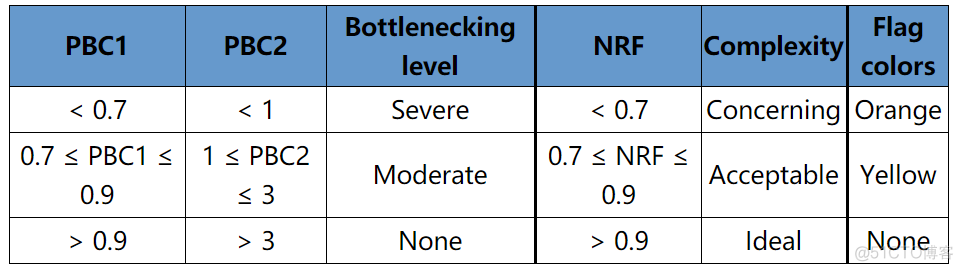
关于文库复杂度的解释可以参考这篇文章
chip_seq质量评估之文库复杂度每个样本的peak数量要在15万以上,10万以上是最低标准;使用IDR评估生物学重复样本的一致性,经过IDR处理后的peak为IDR peak,同时也会给出一个IDR score值, IDR peak的数量要在7万以上,5万是最低标准,IDR score的值应小于2peak结果中要存在NFR和mononucleosome peak区域,NFR peak指的是长度小于1个核小体单位长度的peak区域,1个核小体的DNA长度为146bp, NFR peak长度小于146b;ppmononucleosome peak指的是只跨越了1个核小体的peak, 长度在1个到2个核小体单位长度之间,即146到292bp之间,考虑两个核小peak区域的reads所占比例,即FRIrScore值应该大于0.3, 最低标准是0.2TSS Enrichment soce,称之为TSS富集分数,就是计算所有TSS位点测序深度的平均值,这个数值的大小与所用的参考基因组有关系,不同参考基因组对应的阈值标准如下
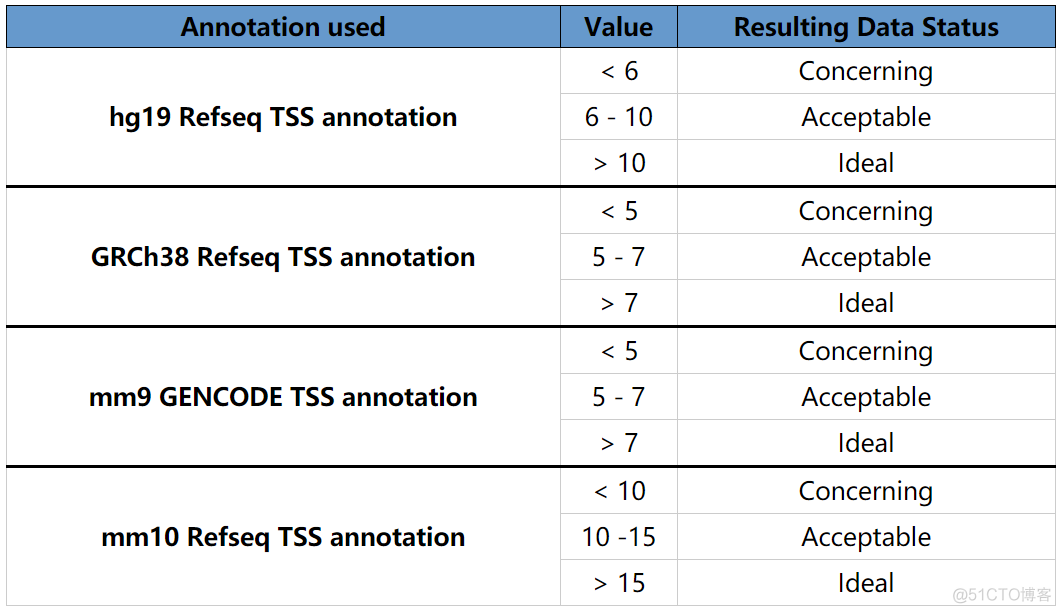
高质量的文库是确保分析结果准确的前提和保障,参考以上几个指标,有助于我们判断ATAC文库的质量。
·end·

一个只分享干货的
生信公众号