除了测序数据量和质量外,ATAC文库还有一些独有的QC指标,比如以下几个指标
为了更好的衡量ATAC的文库质量,有很多的软件相继被开发出来,ATACseqQC是应用的较为广泛的一个。该软件是一个R包,存储在bioconductor上,链接如下
https://bioconductor.org/packages/release/bioc/html/ATACseqQC.html
在这个R包中,提供了丰富的函数,这里只展示常用的几种
1. 插入片段长度分布图
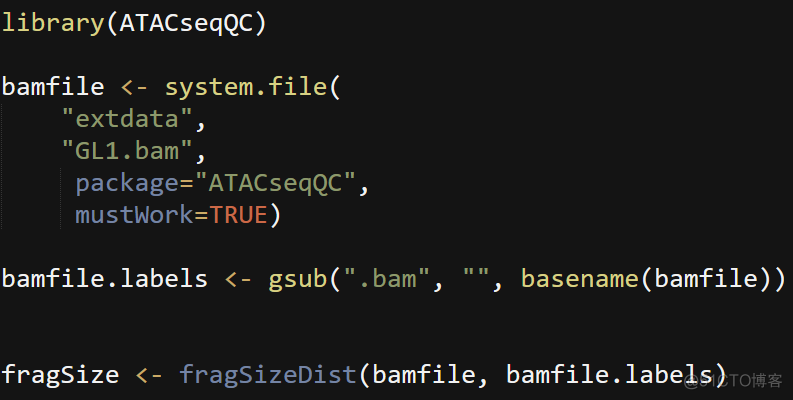
通过fragSizeDist函数可以方便的绘制插入片段长度分布图,代码如下
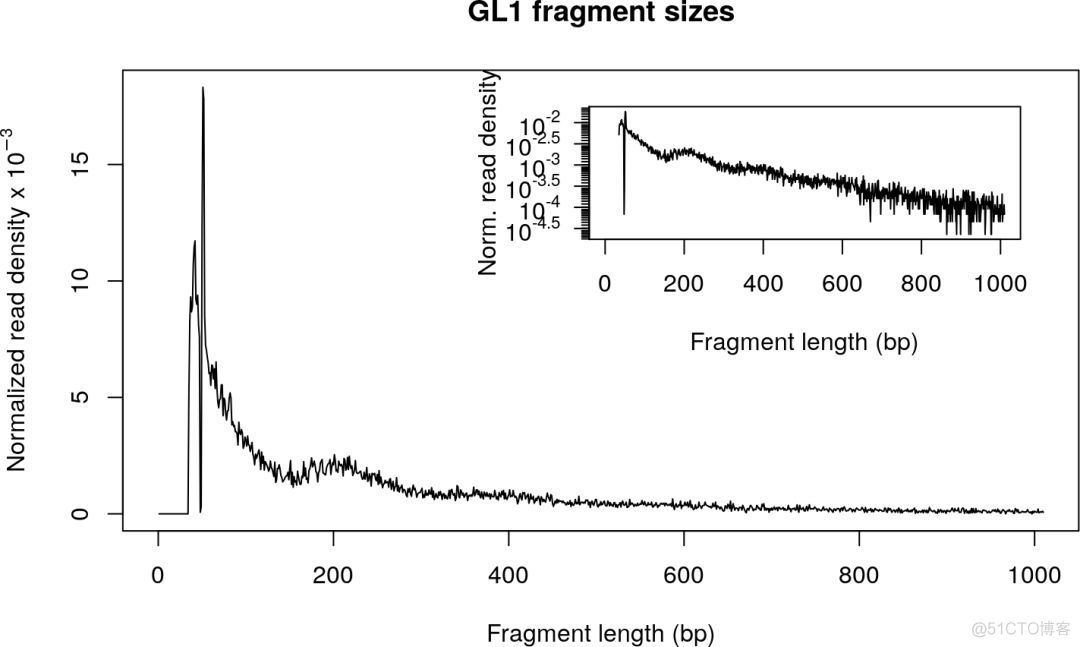
输出结果示意如下
2. shift bam files
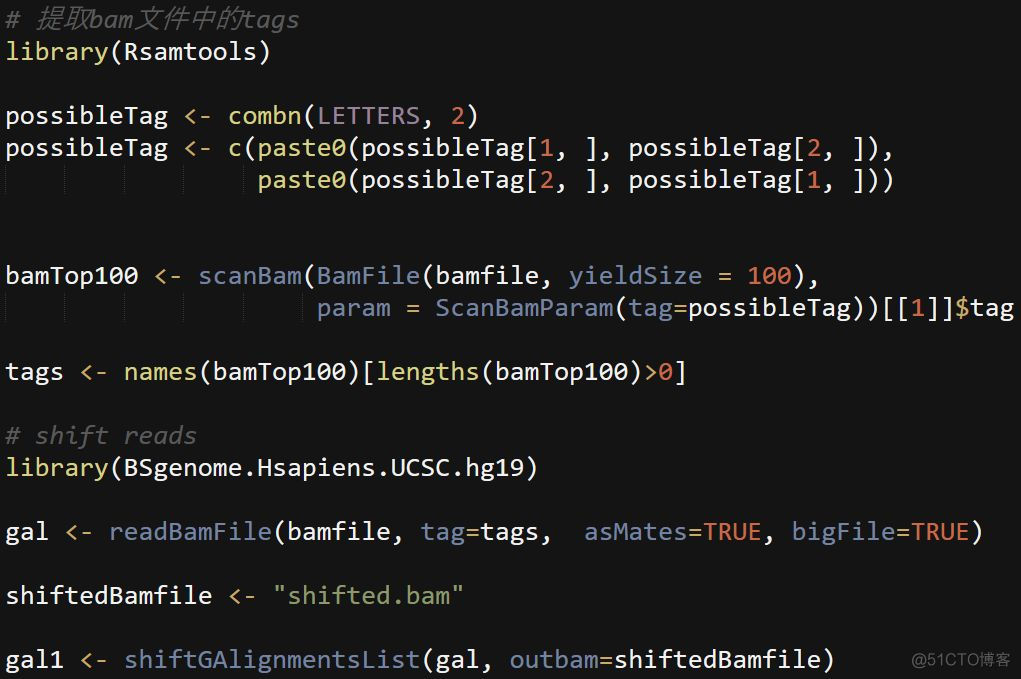
Tn5转座酶切割的序列末端有9bp的gap,在下游peak calling分析之前,需要将bam文件中reads的基因组位置进行偏移,正链向右增加5bp,负链向左减4bp, 对应的代码如下
3. 计算TSS Enrichment scores
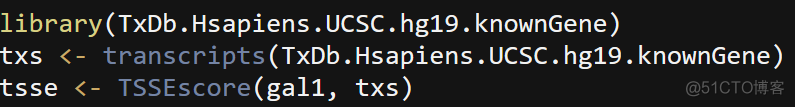
Encode将TSS Enrichment score作为ATAC文库的质控标准之一,计算方式如下
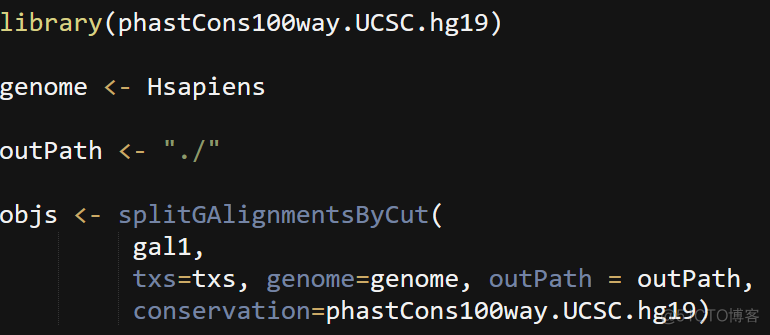
4. split reads
根据reads跨越的核小体个数,ATAC文库中的reads可以划分为NFR, mononucleosome等不同类型的reads, 其中NFR reads在TSS位点两侧显著富集。为了更好的查看NFR reads的分布,需要从bam文件中提取NFR reads, 代码如下
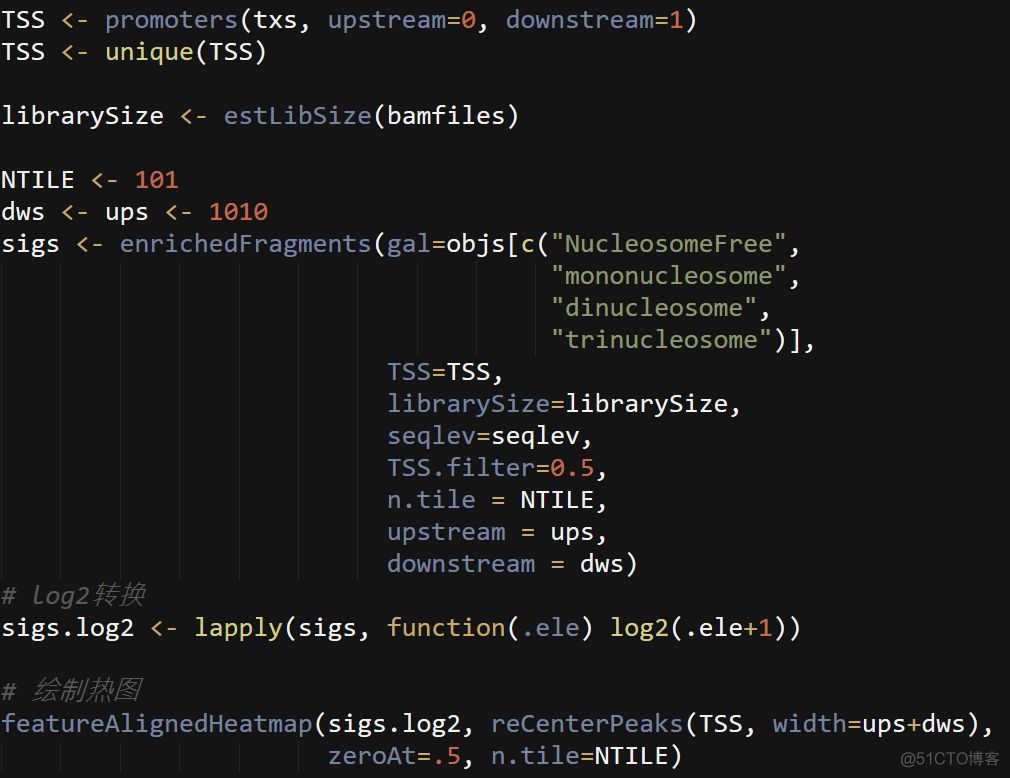
5. TSS位点两侧reads分布图
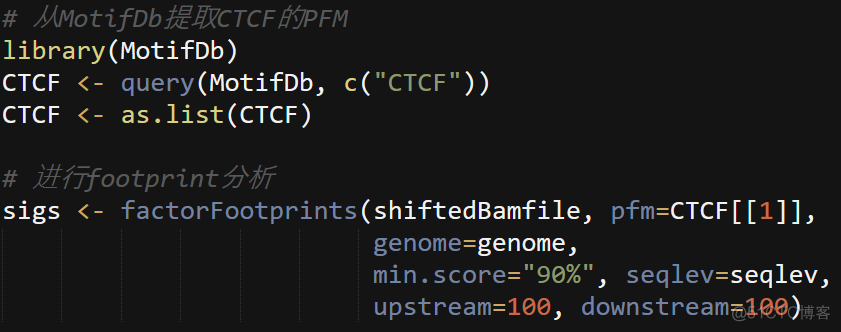
6. footprint
ATAC可以用于寻找正在发挥作用的转录因子,首先根据转录因子的motif序列,在ATAC得到的reads中查找结合区域,然后分析这些结合区域两端reads的分布情况。转录因子结合区域有蛋白保护,是无法被Tn5切割的,只有结合位点两侧的reads会被切割。因此会出现结合区域为空,而两侧区域reads深度高的情况。通过观察特定转录因子结合位点的reads分布,可以判断该转录因子是否正在结合,这个分析称之为footprint分析。代码如下
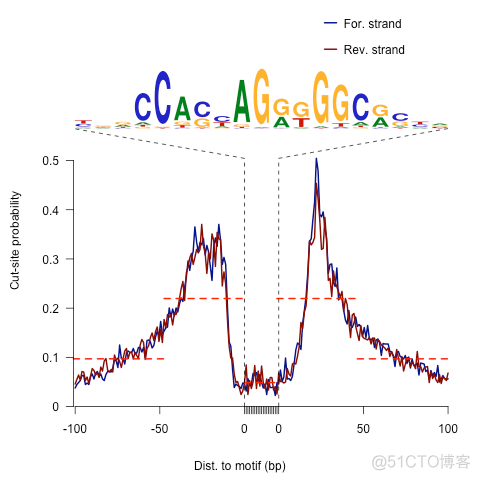
输出结果示意如下
ATACseqQC还提供了非常丰富的功能,详细用法请参考官方文档。
·end·

一个只分享干货的
生信公众号