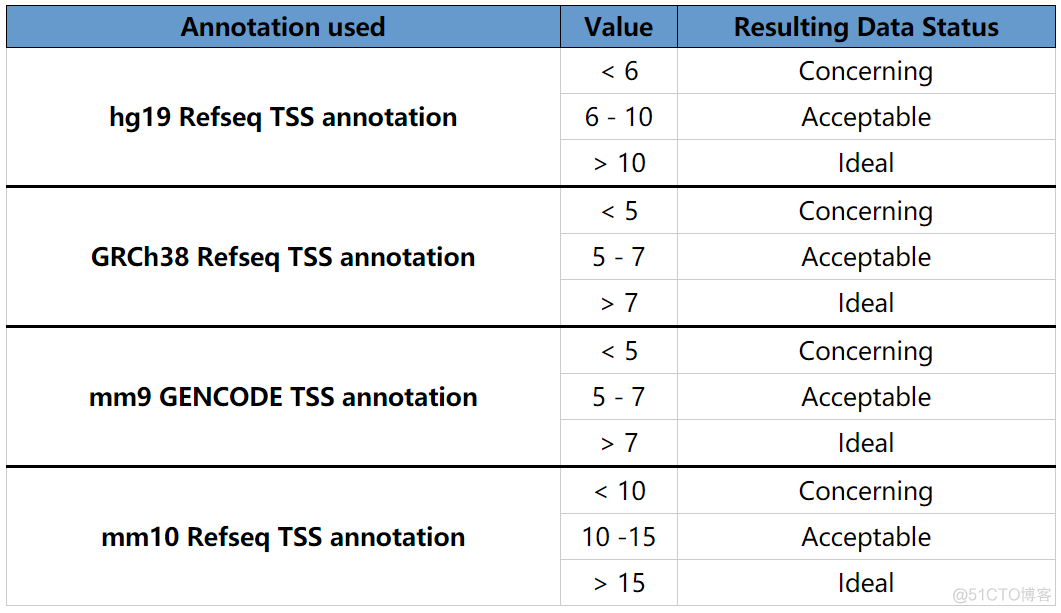
Encode将TSS Enrichment score作为ATAC文库质控的一个指标,不同的参考基因组注释文件,对应的阈值也不同,示意如下
所谓TSS Enrichment score, 其实是所有基因TSS位点测序深度的平均值。要计算这个值,需要两个文件,一个是bam文件,保存了测序深度信息,另外一个是参考基因组TSS位点文件,可以从gtf文件中提取得到,记录了TSS位点的染色体位置。
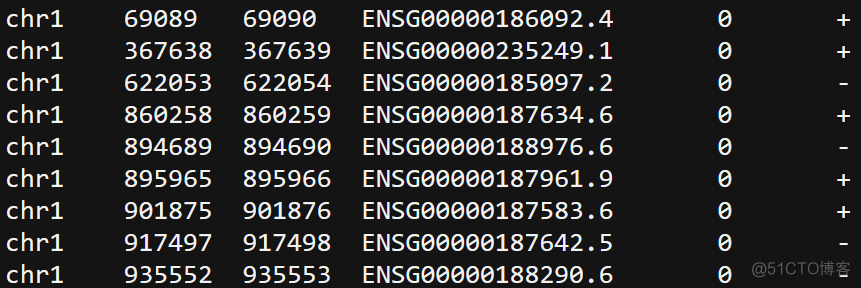
Encode的atac pipeline中提供了人和小鼠的TSS位点数据,以hg19为例,内容如下所示
如果只是粗略计算测序深度的平均值,可以直接利用samtools进行计算,代码如下

更常规的做法是提取TSS两侧区域的reads, 比如上下游各提取2kb的区域,划分等长窗口bin并统计每个窗口内的coverage, 最终会生成一个矩阵,行为基因,列为不同窗口。根据这个矩阵可以绘制TSS两侧reads分布图, 也可以计算TSS Enrichment score。
deeptools就是采用了上述策略,用法如下
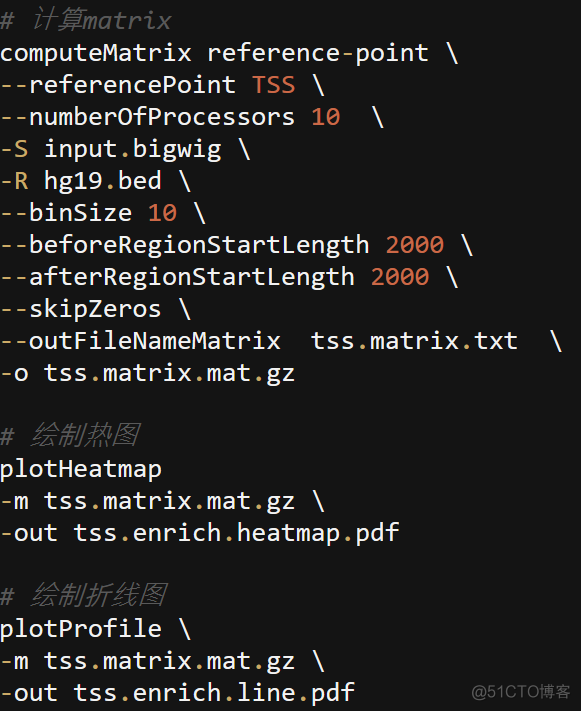
通过deeotools可以直接出图,tss.matrix.txt文件保存了matrix的纯文本信息,可以读取这个文件计算TSS Enrichment score。除比这种方法外,Encode的pipeline还提供了一个脚本,专门用于计算TSS Enrichment score. 链接如下
https://github.com/ENCODE-DCC/atac-seq-pipeline/blob/master/src/encode_task_tss_enrich.py
该脚本采用了pybedtools和metaseq两个模块, pybedtools用于读取TSS bed文件,metaseq读取bam文件,并统计coverage, 该脚本的用法如下
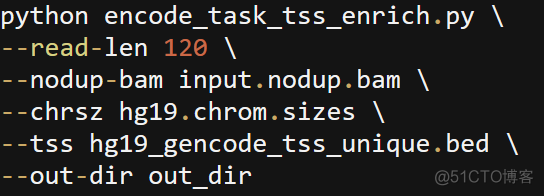
该脚本计算TSS Enrichment score, 并绘制TSS两侧read的分布图。相比deeptools,在计算coverage的过程中进行了归一化,核心代码如下
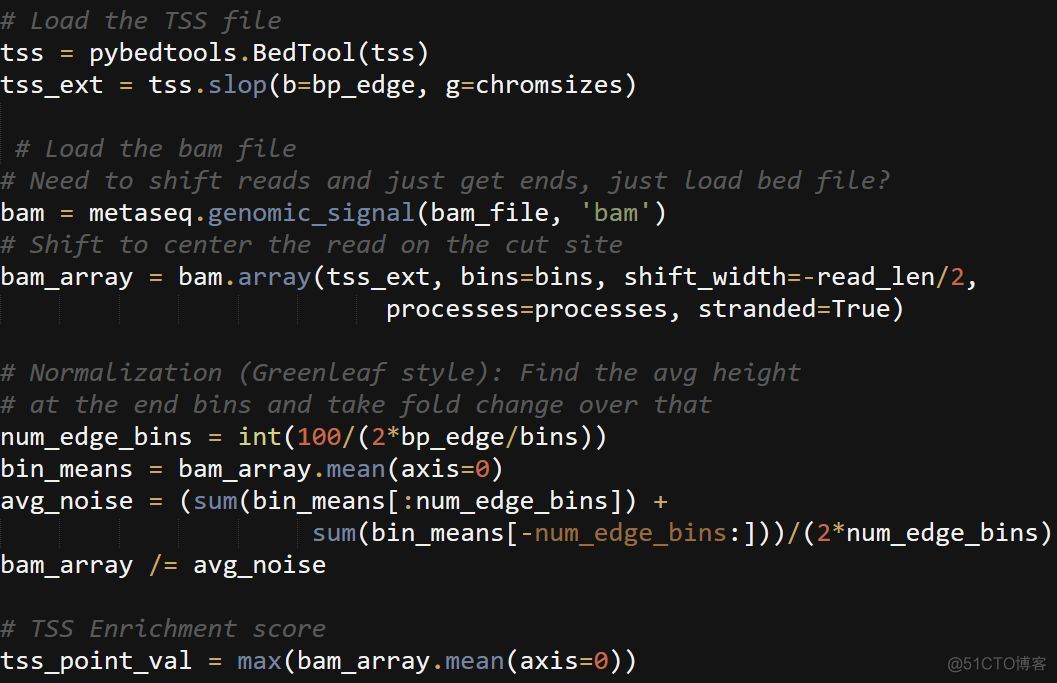
归一化之后,绘制的分布图曲线更加的平滑。上述3种方法计算出的TSS Enrichment score值并不完全一致,更加推荐使用最后一种方法。
·end·

一个只分享干货的
生信公众号