在之前的文章中,我们解读过一篇引用达2000多次的ATAC经典文献
引用2115次的ATAC经典论文解读
这篇文章中,使用了Genrich这个软件来进行peak calling。该软件适用于chip_seq, DNase_seq, ATAC_seq等多种文库的peak calling,源代码保存在github上,链接如下
https://github.com/jsh58/Genrich
该软件用法简单,只需要输入排序之后的bam文件即可,其peak calling的基本算法如下所示
该软件有两种运行模式,第一种适用于chip_seq, 第二种适用于ATAC。对于ATAC的reads,需要对reads比对位置进行shift, 所以peak calling的模式也会有所不同,图示如下
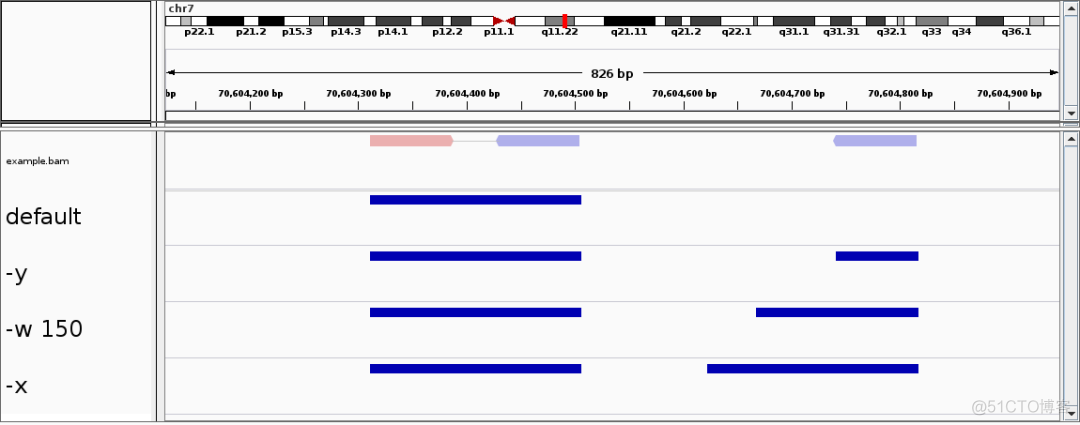
默认情况下为第一种模式,通过-j参数可以调整为ATAC模式。默认模式下,将fragment两个末端对应的区域直接作为peak, 以chip_seq为例,抗体抓下来的reads就是蛋白结合区域的reads。而ATAC文库中Tn5切割的序列是在结合区域的两侧,所以在ATAC模式中,以fragment的中心点为基准,来判断真实的peak区域。
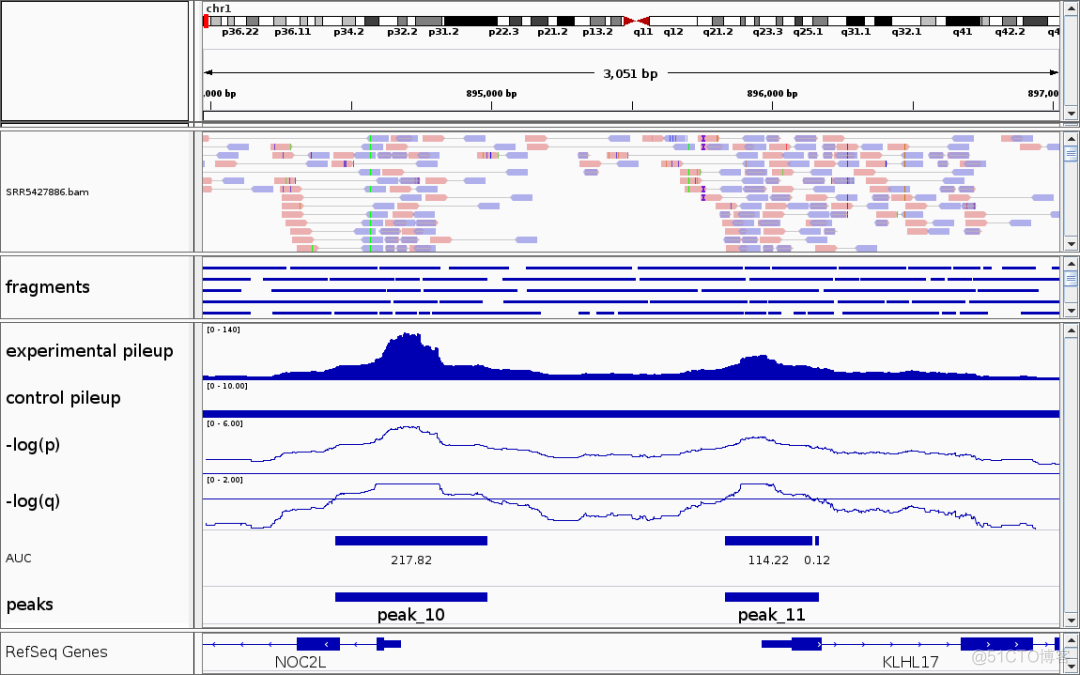
对于ATAC数据的peak calling而言,该软件的用法如下
Genrich \-t SRR5427886.bam \
-o SRR5427886.narrowPeak \
-f SRR5427886.log \
-r \
-x \
-e chrM \
-E wgEncodeDukeMapabilityRegionsExcludable.bed.gz \
-q 0.05 \
-a 20.0
-t参数指定输入的bam文件,-o参数指定输出的peak文件,-f参数指定log文件的名称。-r参数表示去除PCR重复,-x参数表示保留只有一端比对上基因组的reads,-e参数剔除指定染色体上的序列,-E参数剔除bed文件中指定的基因组区域。-q参数指定qvalue的阈值,-a参数指定AUC面积的阈值,
对于ATAC的peak calling而言,除了MACS2, 该软件也是一个不错的选择。
·end·

一个只分享干货的
生信公众号