在Encode的ATAC文库质控标准中,认为一个高质量的文库其FRiP score值应该大于0.03,最低也要大于0.02。FRiP全称如下
Fraction of reads in peaks
表示的是位于peak区域的reads的比例,FRiP score是一个比值,其分子是位于peak区域的reads总数,分母是比对到参考基因组上的reads总数。该值最初在chip_seq文库质控中广泛使用
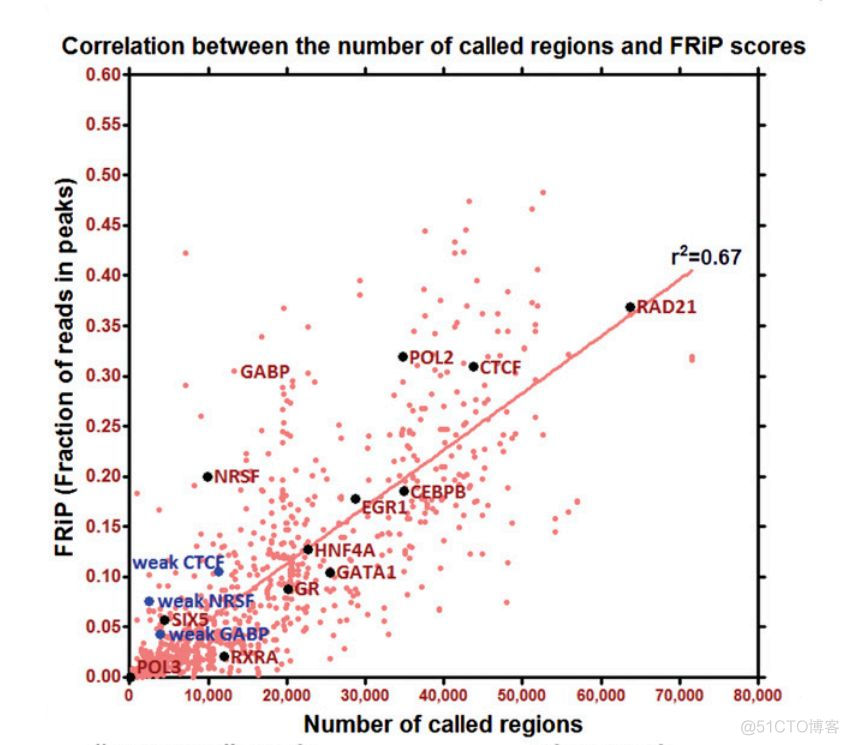
如上图所示,Encode汇总了不同转录因子的chip_seq数据,绘制了peak总数与FRiP score值的分布图。从图中可以看出,FRiP score值与peak 总数呈现正相关关系,而且不同转录因子对应的FRiP score值也不尽相同。
最初制定FRiP score阈值的时候,就是一个经验阈值,观察了上万例样本的FRip score值,发现绝大多数都位于0.01以上,所以采用0.01作为阈值。对于ATAC文库而言,也是同样的思路,由于ATAC的peak总数非常多,所以FRiP的阈值也比较大。需要指出的是,尽管FRiP score的阈值看上去是一个最低标准,但是由于不同组织,细胞类型的特异性,一刀切的标准是很难满足所有情况的。对于不符合FRiP score值的样本,应当结合TSS Enrichment score值等其他指标来进一步衡量其文库质量。
介绍完了FRip Score的概念和应用,我们来看下其计算过程。从概念出发,只需要peak区域内的read总数和mapping上参考基因组的reads总数即可。
在ATAC的peak caling中,使用了TagAlign这种bed文件来存储reads的比对信息,通过这个文件也可以非常快速的计算FRiP score, 步骤如下
1. 计算比对上参考基因组的reads总数
TagAlign格式中,每一行表示一个fragment的比对情况,要统计比对上的reads总数,直接统计行数即可,代码如下
wc -l sample.tn5.shift.tagalign2. 计算peak区域的reads总数
计算peak区域的reads数目,实际上可以转换为peak区域与TagAlign这种bed文件取交集的操作,统计交集的行数即可,代码如下
bedtools intersect -a sample.tn5.shift.tagalign -b sample.narrowpeak -wa -u | wc -l将两个数相除就得到了FRiP score, 需要注意的是,有些人会纠结fragment和read的概念,对于双端测序而言,一个fragment会产生两条reads, 上述的计算过程是针对fragment进行计数的,和reads数目的计算结果是有出入的。在我看来,FRiP score的核心思想是看peak区域的序列占所有比对上基因组序列的比例,用fragment也是对的,没必要精细到read, 而且用fragment可以同时使用单端和双端测序。
·end·

一个只分享干货的
生信公众号