由于Tn5转座酶的特性,在ATAC数据分析中,首选需要对bam文件中reads的比对位置进行shift, 然后再进行peak calling。那么如何进行这一操作呢?直接修改bam文件中reads的比对区域吗?
当然你可以这样操作,但是bam文件的读写是一个非常费时的操作,因为bam文件中包含了序列,比对位置等完整信息,文件非常大。对于下游分析而言,其核心信息是reads比对到参考基因组上的位置,就是坐标,我们只需要提取这个坐标,然后进行shift操作就可以了,此时可以借助TagAlign这一格式来操作,更加简单方便。
首先来了解下什么是TagAlign格式。在使用macs进行peak calling时,除了输入样本对应的BAM/SAM文件之外,还可以输入BED文件。BAM文件我们都非常熟悉,将序列比对到基因组之后就可以产生这样的文件,各个比对软件也支持输出BAM/SAM格式。这种格式的文件记录了序列的比对情况,根据这个文件可以计算出基因组上的测序深度分布,从而比较不同样本的分布进行peak calling, 那么BED文件又是怎么一回事呢?
在BAM文件中,最核心的信息是序列和基因组区域的对应关系,即那些序列比对上了基因组上的哪些区域,这个信息通过BED格式也是可以来记录的。在bedtools中也提供了bamtobed的功能,基本用法如下
bedtools bamtobed -i input.bam > out.bed输出内容示意如下
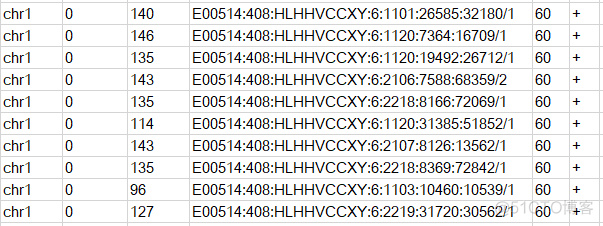
前三列表示reads比对上的染色体位置,第四列为reads的名称,第五列代表比对的质量值MAPQ,第六列代表正负链信息。
这种6列的BED文件在ENCODE被命名为tagAlign格式,详细解释参见如下链接
https://genome.ucsc.edu/FAQ/FAQformat.html#format13
对于双端测序的数据,还有一个特殊的bed格式-bedpe, 用法如下
bedtools bamtobed -i input.bam -bedpe > out.bed内容示意如下
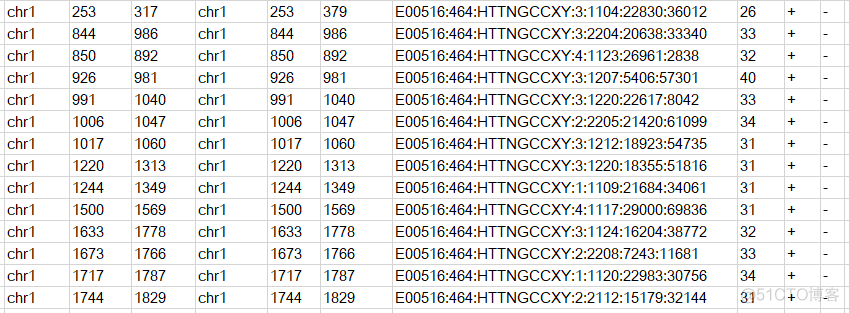
bedpe格式在一行中显示了R1和R2两个reads的比对情况,列数为10列。
对于单端序列。直接用bed格式就可以;对于双端序列,推荐用bedpe格式。这两种格式都可以称之为tagAlign,可以作为macs的输入文件。
tagAligen格式相比bam,文件大小会小很多,更加方便文件的读取。在转换得到tagAlign格式之后,我们就可以很容易的将坐标进行偏移,代码如下
zcat sample.tagAlign.gz | \awk -F '\t' 'BEGIN {OFS = FS}{ \
if ($6 == "+") {$2 = $2 + 4} \
else if ($6 == "-") {$3 = $3 - 5} \
print $0}' | \
gzip -nc sample.tn5.shitf.tagAlign.gz
用偏移之后的文件进行peak calling即可。代码如下
macs2 callpeak \-t sample.tn5.shitf.tagAlign.gz \
-f BED \
--nomodel \
--shift -75 \
--extsize 150 \
-B \
-n sample \
-g hs
在Encode的ATAC分析pipeline中,就是采用上述方法进行reads的偏移和peak calling操作的。
·end·

一个只分享干货的
生信公众号