meta-analysis对多个独立研究的成果进行综合评估,该技术在医学,心理学等领域早已广泛使用。虽然该技术的理论基础早已成熟,但是在GWAS分析领域,还是有很多困难需要去克服
为了有效的进行分析,我们需要使用成熟软件,本文要介绍的GWAMA就是一款很不错的软件,其运行效率特别高,适合针对大数量的SNP位点进行分析,对应的文章发表在BMC Bioinformatics上,链接如下
https://bmcbioinformatics.biomedcentral.com/track/pdf/10.1186/1471-2105-11-288
官方网址如下
https://www.geenivaramu.ee/en/tools/gwama
安装过程如下
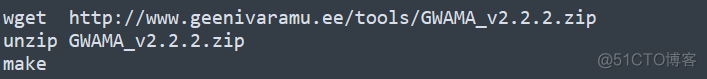
采用C++进行开发,下载源代码解压,编译即可。官网也提供了配套的测试数据,可以用于测试该软件
该软件的基本用法如下
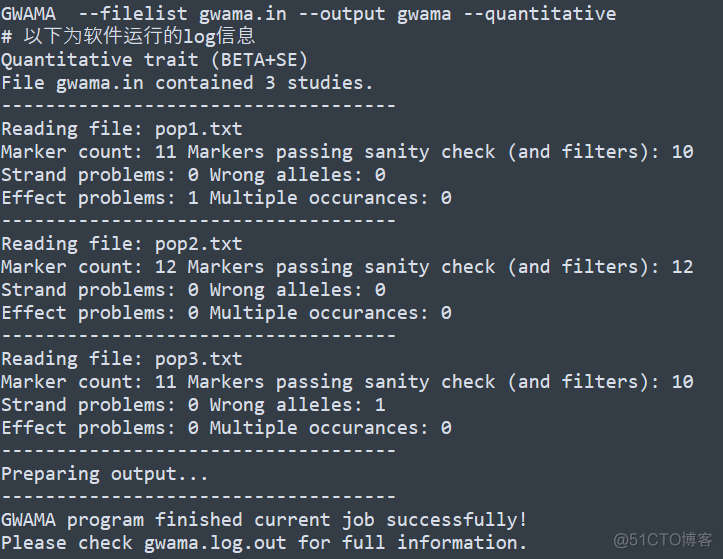
只需要一个输入文件,默认文件名称为gwama.in, 内容示意如下

每一行代表一个study的gwas分析结果,对应的文件格式如下
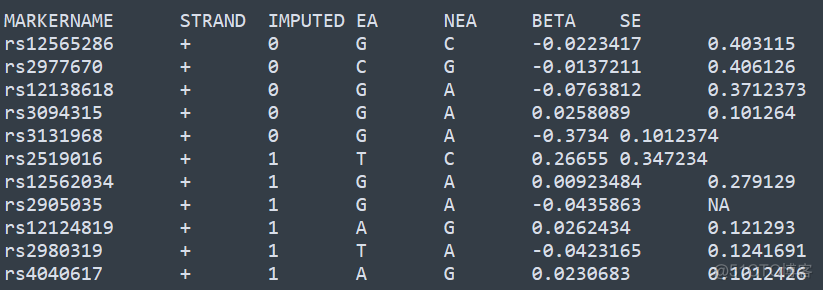
MARKERNAME代表SNP位点的名称,STRAND代表链的方向,IMPUTED代表该位点是否是填充得到的,EA代表tested allele, NEA代表other allele, 对于连续型的性状,要求输入BETA和SE两列,对于二分类的性状,要求输入OR, OR_95L, OR_95U3列,代表odd ratio以及95%置信区间。
默认输出结果保存在gwama.out文件中,列数较多,为了方便展示,我这里截取了部分并进行了转置,内容如下
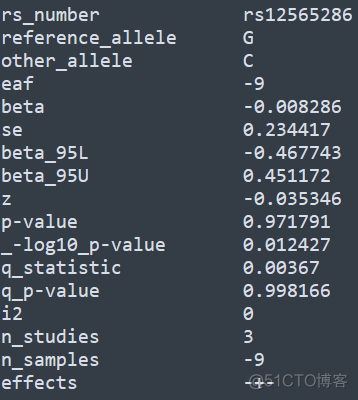
另外该软件还支持针对性别进行校正,更多用法请查看官方文档。
·end·

生物信息入门
只差这一个
公众号