CNVnator是一款利用全基因组数据进行CNV检测的软件,对应的文章链接如下
https://genome.cshlp.org/content/21/6/974.long
基于read depth的分析策略, 采用滑动窗口的方式,将基因组划分为等长的窗口,称之为bin, 利用不同窗口内测序深度的分布来预测CNV, 如下图所示
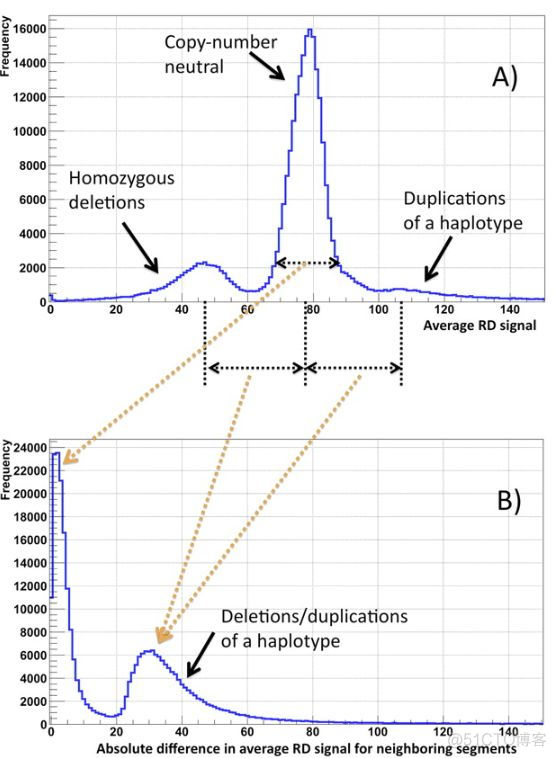
图A中横坐标为平均测序深度,用RD signal表示,纵坐标为对应的bin的频率,实际上是不同测序深度频率分布图,在图中可以看到三个峰,最高的一个拷贝数正常的染色体区域对应的bin, 另外两个峰分别表示纯合缺失和单体型重复。图B表示与邻近区域的测序深度差异值的分布,不同的峰对应不同的拷贝数情况。
从上图可以看出,利用测序深度的分布能够反映出染色体区域的拷贝数差异。cnvnator软件的算法具体可以分为以下几步
1.比对参考基因组
要计算测序深度,首先需要将测序的reads比对到参考基因组上,比对是最关键的一个步骤就是如何比对到基因组多个区域的reads。当一条reads比对到基因组上的多个位置时,单从数据分析的角度,是完全无法区分其究竟属于哪一个区域的,因为这些区域同源度非常的高。对于这样的reads, 有两种处理策略,第一种是直接剔除,保留unque-mapping的reads; 第二种是随机选取其中的一个位置,作为该reads的真实比对位置,cnvnator算法采用的是第二种策略。
2. 构建RD signal
比对之后,就可以将基因组划分为等长窗口,计算每个窗口内的测序深度了,这里需要注意的是, 利用gc含量在校正原始的测序深度。PCR对不同GC含量序列的扩增存在偏倚,所以在计算窗口内的RD signal, 需要校正这一系统误差,cnvnator的校正公式如下
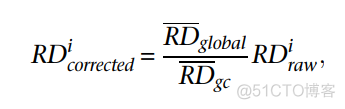
global表示所有bin窗口内原始RD signal的平均值,gc表示和当前bin的GC含量相同的所有bin窗口原始RD signal的平均值,将二者的比值作为一个系数,对原始的RD signal进行校正。
3. mean-shift 聚类
mean-shift是一种聚类算法,利用校正之后的RD signal值,对邻近的bin进行聚类,理论上聚为一类的bin具有相同的cnv拷贝数,图示如下
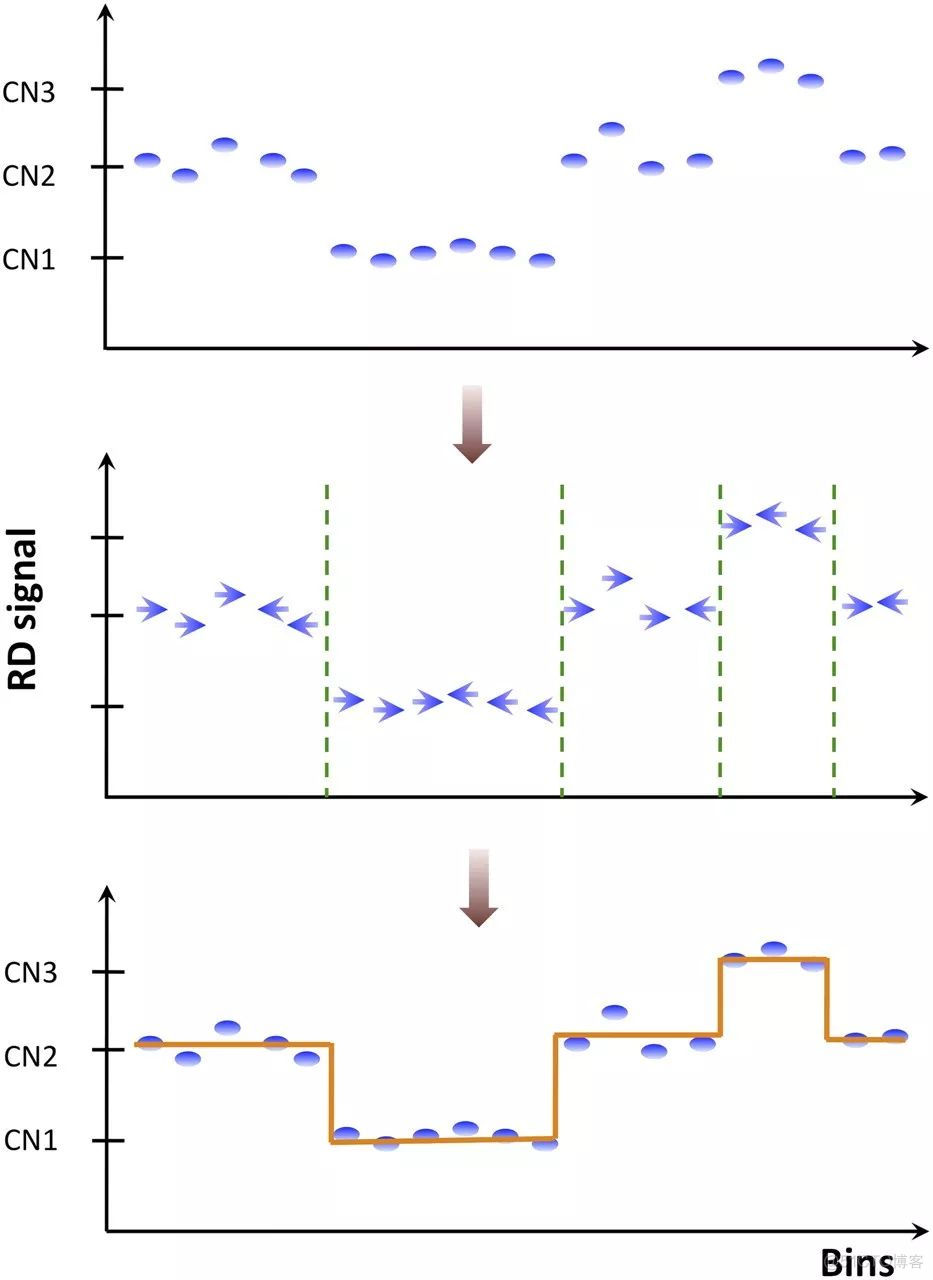
需要注意的是,这里只是对染色体位置接近的bin进行聚类,并不是等同于CNV分析中的segmentation。
4. segmentation
上述的聚类信号只有在染色体的局部具有意义,当放到大全基因组范围来识别CNV时,必须通过segmentation算法来实现,cnvnator采用的是自己独特的算法,有个关键的参数称之为bandwidth, 不同的取值会影响到CNV区域的划分,图示如下
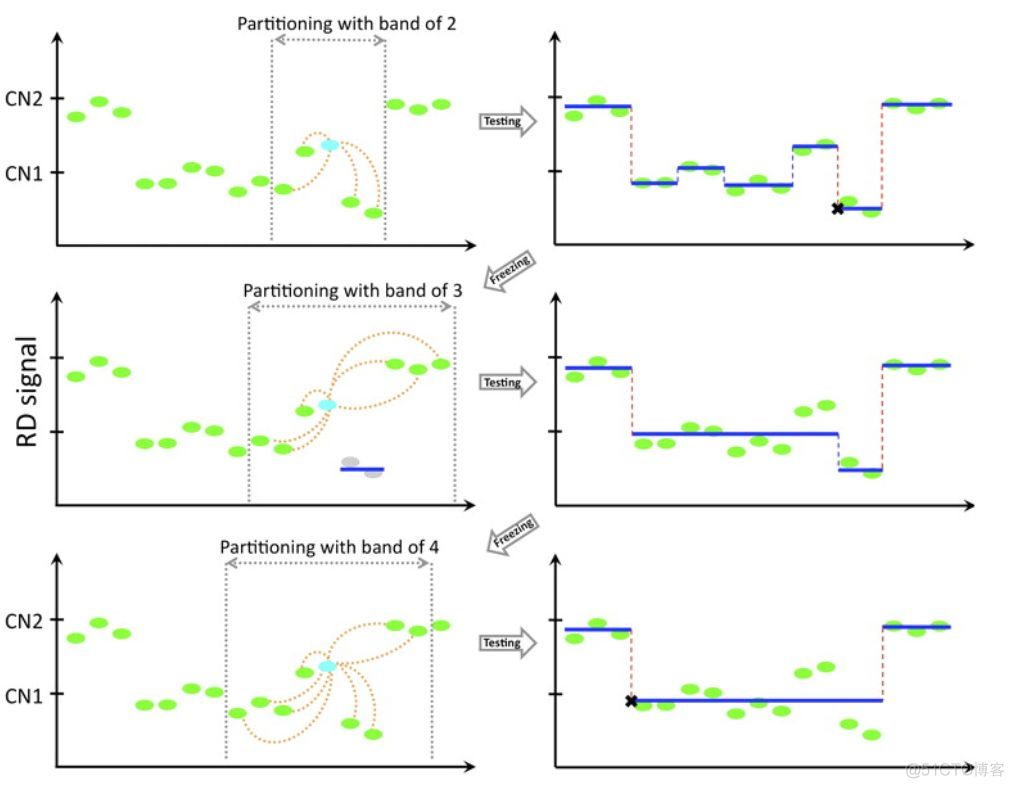
取值越大,小片段的CNV信号会被掩盖,取值越小,数值越小,CNV检测的假阳性率会高。
5. signal merging
根据与邻近segment RD signal的差异, 将原始划分的segment进行合并。
6. cnv calling
对划分好的不同segment, 预测其对应的拷贝数。
在利用CNVnator软件进行分析时,bin和bandwidth两个参数的选择对结果影响很大。通过该软件可以检测各种长度的cnv, 而且分型的准确率非常高,是一款值得推荐的cnv检测软件。
·end·
