作为一种经典的可视化方式,曼哈顿图使用广泛,在GWAS分析中随处可见,本文就来揭秘曼哈顿图绘制的核心方法。
曼哈顿图的命名得益于其形状,和纽约市曼哈顿区鳞次栉比的大楼非常相近,曼哈顿区是摩天大楼最多的城市,标志性的景观如下

曼哈顿图示意如下
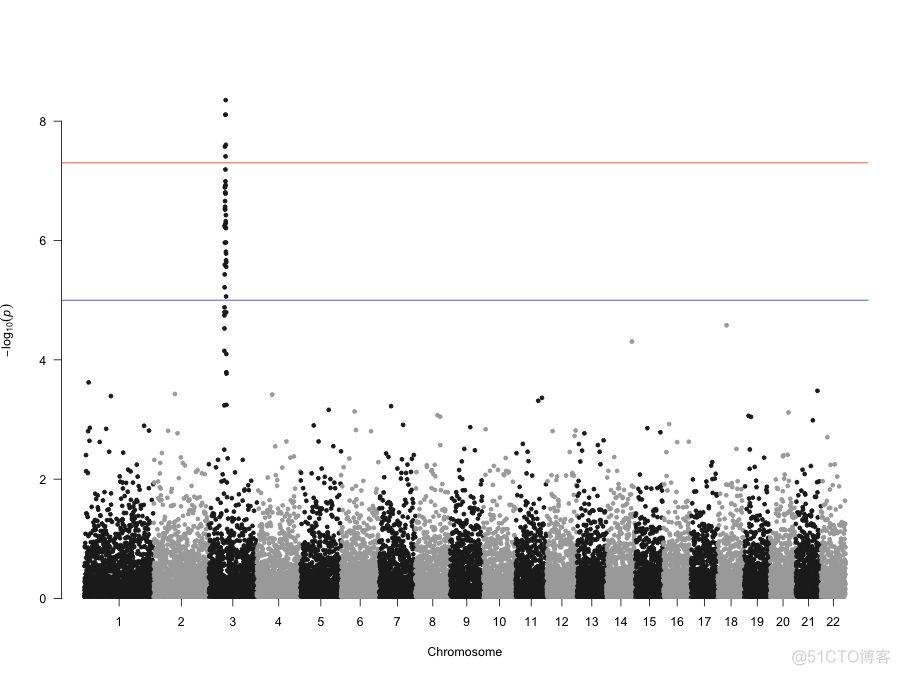
每条染色体可以看做是一座高楼,整体看上去形似曼哈顿区的摩天大楼,所以称之为曼哈顿图。
了解了曼哈顿图的命名,再来看下它所展示的信息。从上图可以看出,曼哈顿图的x轴为snp位点在染色体上的位置,y轴为SNP位点对应的p值。从本质上来看,属于散点图。有x轴和y轴的坐标,人人都可以画散点图,那为何曼哈顿图的绘制看上去并非如此简单呢?
秘密就在于SNP位点的染色体位置,对于不同的染色体,在标记位置时都是从1开始计数的,比如1号和2号染色体的12bp对应的是两个snp位点。通过染色体和位置两列信息相结合, 可以唯一表征snp位点的位置。
然而在绘制散点图时,x轴只能用1个数值来表示。在曼哈顿图中,将染色体线性排列,1号染色体的位置不变,2号染色体的位置在原来的基础上再加上的1号染色体的总长度,然后依次类推,通过这样一种形式,将原始输入文件中染色体和位置两列信息,转换为绘图所用的x轴坐标信息。
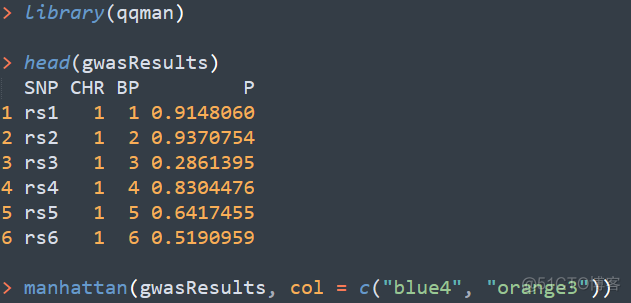
在实际分析中,通过qqman这个R包可以来实现曼哈顿图的绘制,用法如下
输出结果如下所示
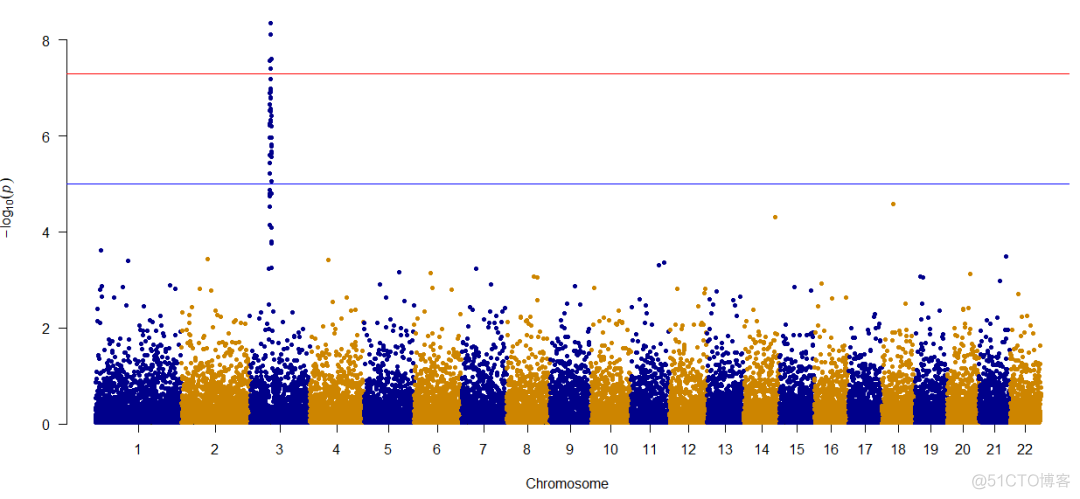
可以看到,只需要准备好同样格式的输入文件,绘制曼哈顿图就是几秒钟的事情。曼哈顿图展示了每个SNP位点的p值信息,图中的两个水平横线对应筛选显著位点的阈值,一个对应的p值是1e-5,另一个对应的p值是5e-8。
当然该R包还提供了很多的功能,比如高亮标记指定的snp位点等,改变染色体显示的名称等等,能够满足基本需求,但是还是由许多限制,比如不能展示性染色体。该包的源代码中会自动对数据过滤,只保留常染色体上的SNP位点。
使用别人的R包就是这个样子,别人给你什么,你用什么,想要个性化,要么通知R包的开发者新增功能,当然人家不一定会买账,要么自己来。
将qqman中的核心源代码部分截取出来,如下
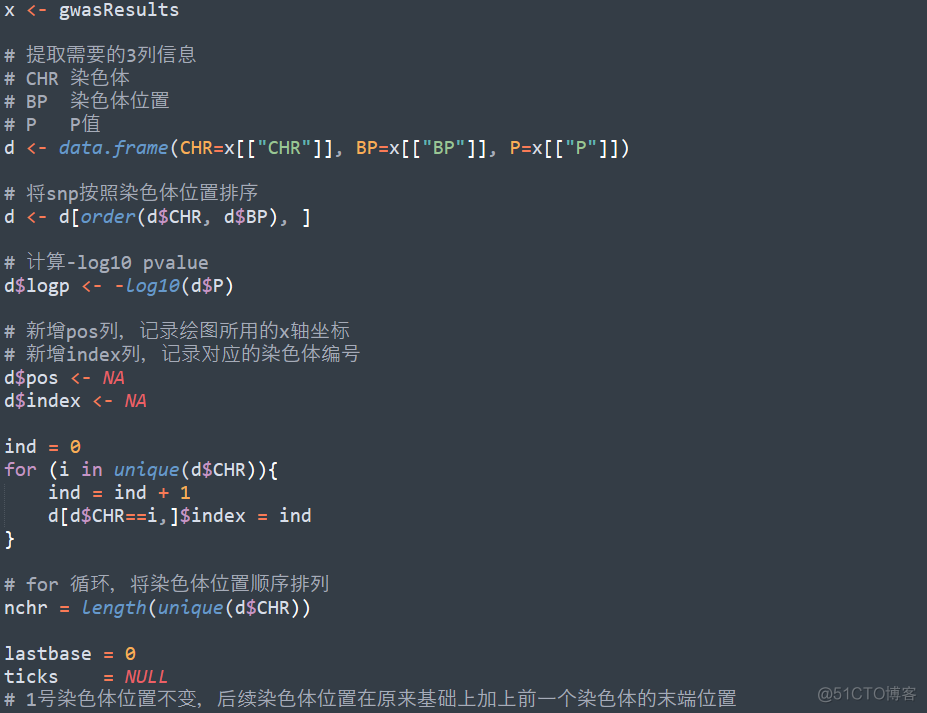
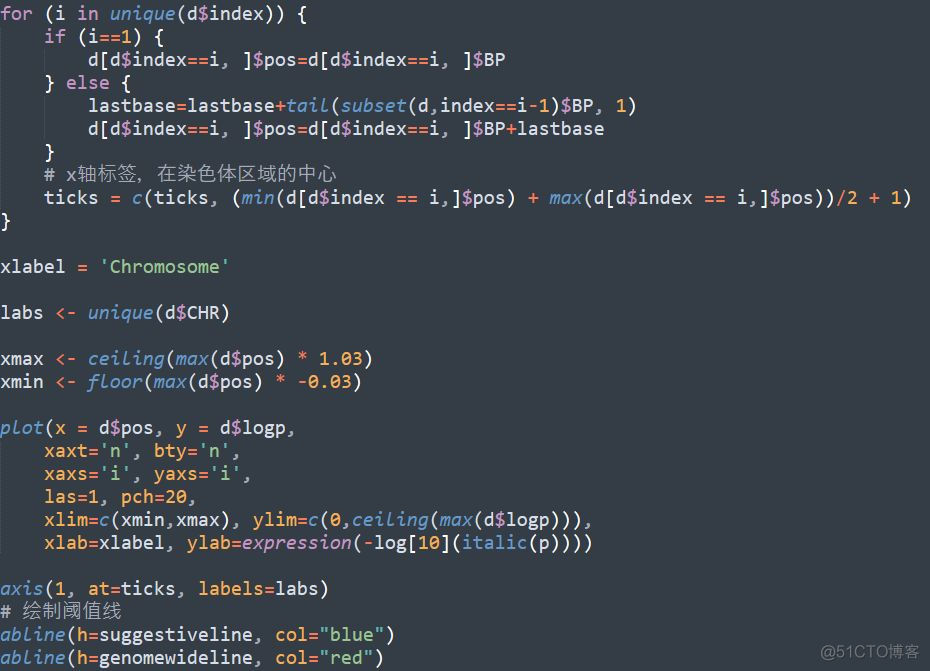
这里省略了颜色的赋值,可以看到,大部分的工作是在计算snp位点对应的x轴坐标,画图的代码只有寥寥几句。
理解了曼哈顿图的本质,就可以自已用R或者熟悉的软件来定制曼哈顿图。
·end·
