对于chip_seq, atac_seq等实验而言,生物学重复样本的peak calling结果很难完全一致。对于多个生物学重复样本的peak calling结果, 如何筛选出最终的可以代表这一组样本的peak是一个难题。
目前常见的策略有以下几种
IDR是Irreproducible Discovery Rata的缩写,代表不可重复性率,是一个专门用于从多个生物学重复样本的peak结果中提取高一致性peak区间的软件,源代码托管在github上,网址如下
https://github.com/nboley/idr
传统分析中,常常采用斯皮尔曼相关性来衡量生物学重复样本的一致性, 比如RNA_seq, 首选去除低丰度的基因,然后计算相关性。之所以要去除低丰度,是由于低丰度的定量结果更可能是噪音,而不是真实的信号,通过一个经验阈值来区分噪声和真实的信号。这种方法依赖阈值,不同的阈值结论也会有差异,而且只考虑了数值的排序,没有考虑数值的差异。
在IDR软件中,摒弃了这种用经验阈值来区分signal和noise的方法,直接输入全部的结果即可,软件会自动根据在生物学重复样本中的分布来确定合适的阈值,所以要强调一点,对于IDR的输入文件,事先不需要做任何过滤和筛选,直接使用最原始的peak calling结果即可。
将signal和noise区分开之后,进一步将signal分成reproducible和inreproducible 两类, 默认情况下只选取存在overlap的peak进行分析, 首先对其排序,排序的依据可以是fold enrichment, pvalui或者qvalue,这个参数可以调整,将所有信号排序之后给每个信号赋值一个IDR value, 来衡量这个信号在生物重复样本中的一致性,数值越大,不可重复性越高。最终根据IDR value的阈值,筛选小于阈值的peak即可。
IDR软件的算法对数据的分布没有任何先验假设,适用范围广泛,Encode在其官方流程中也适用这个软件来处理生物学重复的peak。该软件用法也非常简单,基本用法如下
idr --samples peak1 peak2 --peak-list merge.peak --plot最基本的输入文件为每个生物学重复样本的peak calling结果, 用samples参数来存储,多个样本的结果用空格分隔,除此之外,还可以提供生物学样本合并之后的peak calling结果,用peak-list参数来调整,--plot参数对IDR value, rank的分布等信息进行可视化。结果示意如下
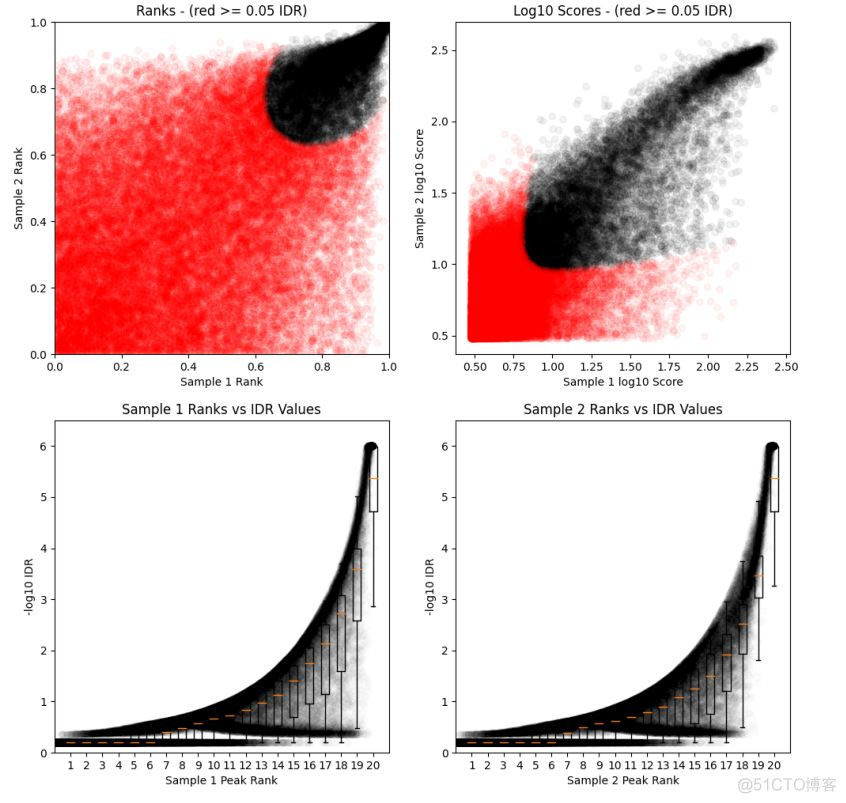
默认情况下统计IDR < 0.05的peak, 这个阈值可以通过soft-idr-threshold参数来调整。在输出文件中,保存的是所有peak的结果,需要自己通过IDR value的值来进行筛选,输出文件的第12列记录了peak对应的global IDR value的值,通过这个值进行筛选即可。
通过IDR软件可以很方便的处理生物学重复样本的peak calling结果,筛选出一组一致性高的peak。
·end·

一个只分享干货的
生信公众号