基因结构是最基本的基因组注释信息,通常情况下,我们最关心基因区域内的数据分布情况,有多种文件格式可以存储基因结构信息
用固定格式来存储对应的信息,使得生物信息软件可以更加标准化其输入输出,为数据分析带来便利。但是存储在文件中的信息对于我们而言,并不够直观。为了更加直观的查看基因结构,可以使用IGV浏览器,只需要将对应格式的文件导入软件中即可。
基因结构信息的本质是染色体坐标,IGV要求导入的数据必须是排序之后的结果。以GTF文件为例,可以采用如下命令先进行排序
sort -k1,1 -k4,4n -k5,5n hg19.gtf > hg19.sort.gtf排序之后还需要对文件建立索引,这样检索的速度会更快,用igvtools可以建立索引,命令如下
igvtools index hg19.sort.gtf运行完成后,会生成一个后缀为idx的文件,将排序后的gtf文件和其索引放在同一个目录下,然后导入gtf文件即可。导入成功之后, 可以看到如下所示的结果

所有的转录本折叠在同一行进行展示,下方是对应的gene name。这种展示方式称之为Collapsed, 比较节省空间,但是很多的转录本折叠在一起,无法相互区分。
同一个基因的多个转录本会存在重叠,相邻基因的转录本也可能存在重叠,为了更加的区分重叠的转录本,还支持以下两种展示方式
1. Expanded
结果示意如下

2. Squished
结果示意如下
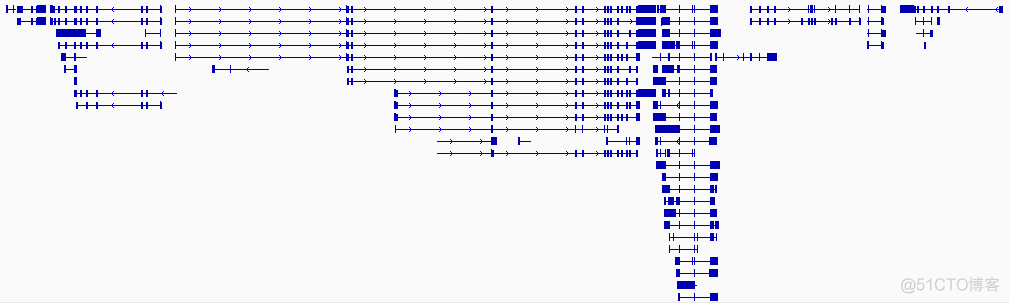
通过右键可以切换不同的展示方式,Expanded模式下转录本区分的最清楚,但是占据的空间很大,Squished则是一种折中方案,抛弃了gene_name, 进一步压缩了空间。
每一条转录本,由3种元素构成
示意如下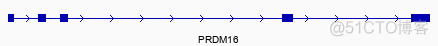
其中矩形表示exon区域 ,线条表示基因的正负链信息,向右的箭头表示正链,向左的箭头表示负链。有时会看到类似下图的转录本结构

上图中较窄的矩形区域表示的是UTR区域,对于蛋白编码RNA, 当GTF文件中提供了UTR或者CDS的区间时,会自动计算出UTR区域并进行标注。
·end·

一个只分享干货的
生信公众号