将bam文件导入IGV之后,可以直观的查看测序深度的分布情况, 但是直接导入bam文件会占用比较大的内存,如果只是想要查看测序深度信息,有很多其他的代替方案。
tdf是IGV官方推荐的一种二进制格式,类似bedgraph格式,用窗口的方式来记录测序深度信息。相比bam文件,tdf文件会小很多,导入和查看也更快速。可以通过igvtools来生成tdf文件,命令如下
igvtools count input.bam out.tdf hg19.chrom.sizes需要三个参数,第一个参数为输入文件,支持bam, sam等格式,第二个参数为输出文件,支持tdf和wig两种格式,第三个参数为基因组的ID或者保存染色体大小的chrom.sizes文件。在igvtools的安装目录,有个lib/genomes文件夹,保存了很多物种的chrom.sizes文件
当你提供了基因组ID时,软件会自动在该目录下查找对应的文件,默认的ID是hg18, 当然你也可以自己准备这个文件,内容如下所示
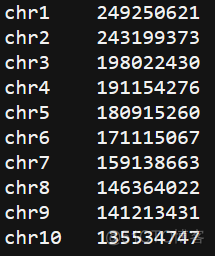
\t分隔的两列,第一列为染色体名称,第二列为染色体长度。在我的测试中,bam文件大小为1.3G, 输出的tdf文件大小为17M, 而wig文件为59M,转换为二进制的bigwig文件大小为25M。可以看到,tdf的文件大小最小。输出的tdf文件可以直接导入IGV进行查看,示意如下
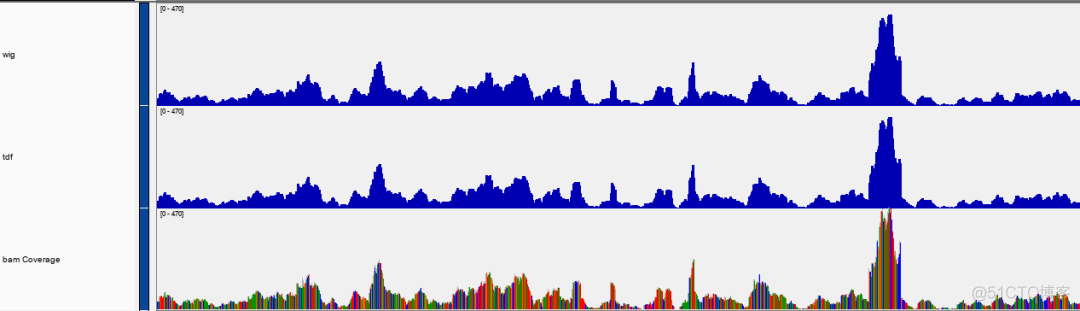
上图的wig,tdf来自同一个bam文件,从峰型可以看出,三者基本一致。只所以说基本一致,是因为wig和tdf都是以窗口的方式来统计测序深度的,而bam则以单个碱基为单位。在IGV中进一步放大,可以看到下图
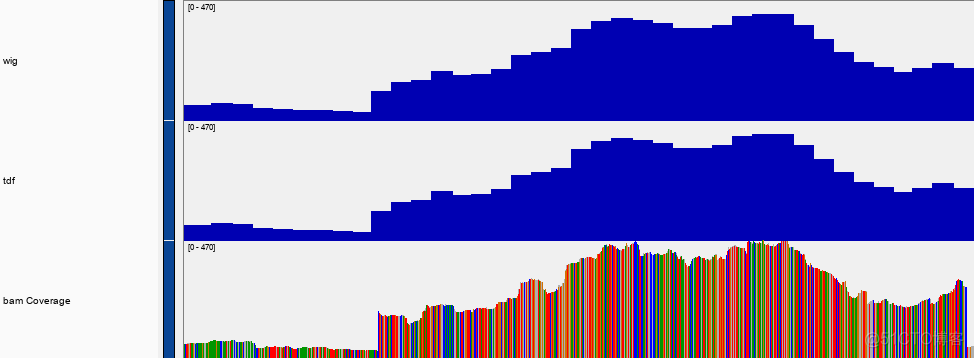
wig和tdf不再是平滑的峰,变成了一个个很宽的柱子。相比之下,bam文件依然是以碱基为单位的柱子。通过下面的命令,可以将tdf文件转换为bedgraph这种纯文本格式
igvtools tdftobedgraph input.tdf out.bedgraph内容如下
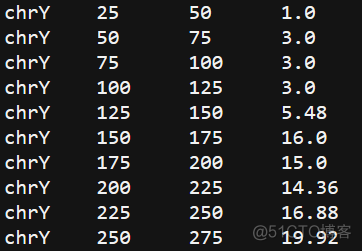
可以看到,tdf默认以25bp为窗口,统计该窗口内测序深度的平均值。窗口大小是可以调整的,可以通过如下命令进行调节
igvtools count -w 50 input.bam out.tdf hg19.chrom.sizestdf和bigwig两种格式都是以窗口方式统计测序深度,窗口越大,对应的文件大小越小,而分辨率也越低。
·end·

一个只分享干货的
生信公众号