后缀为cel的芯片文件,对应的芯片平台为Affymetrix, 针对这一平台的数据,可以通过R包affy来读取,读取时我们需要以下两种文件
1. 后缀为cel的探针荧光信号强度文件
2. 后缀为cdf的探针布局文件
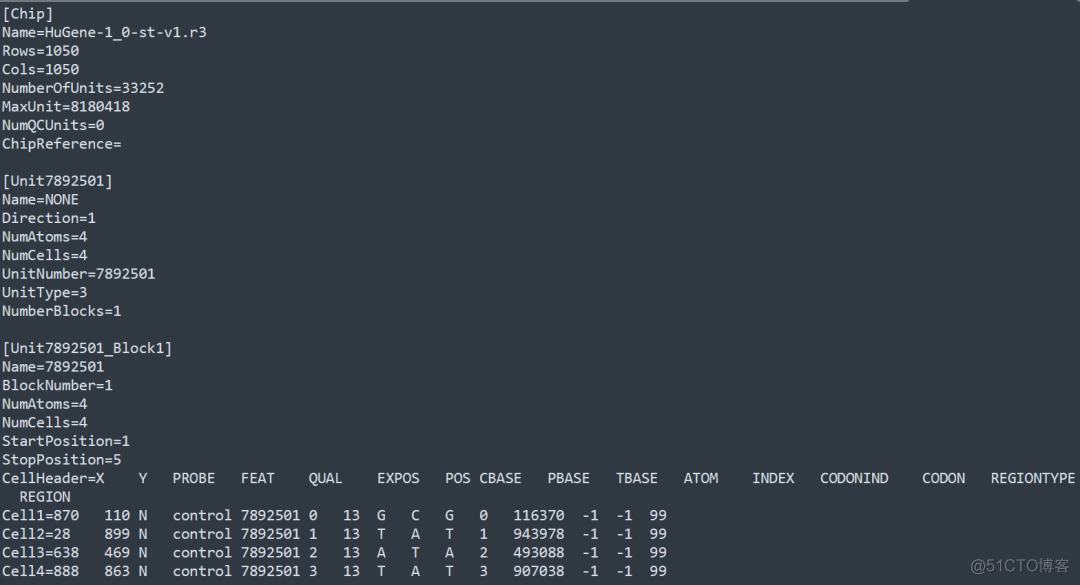
cel文件是芯片扫描之后的原始数据文件,而cdf文件是每个芯片平台对应的文件,cdf格式的详细解释可以参考如下链接
https://media.affymetrix.com/support/developer/powertools/changelog/gcos-agcc/cdf.html
部分内容截图如下
针对一些常用芯片, bionconductor annotation收录了对应的cdf注释信息,链接如下
http://master.bioconductor.org/packages/release/data/annotation/
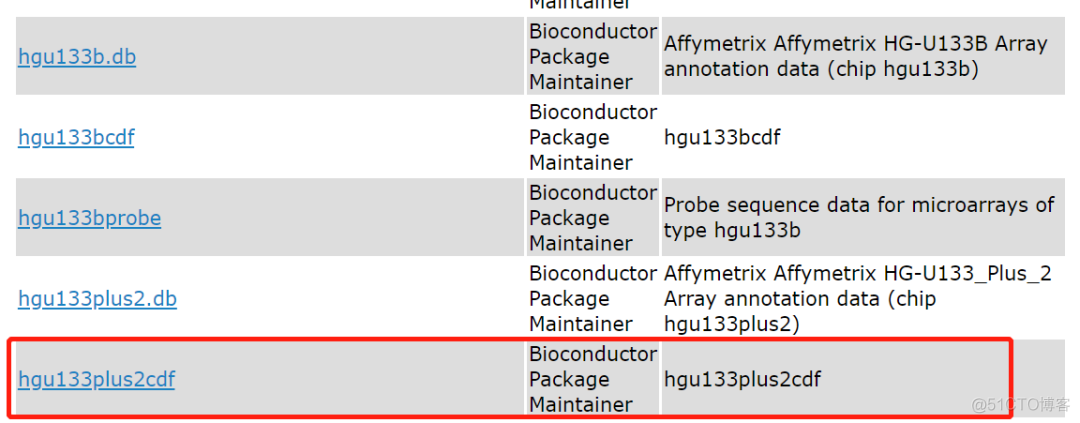
在读取数据的过程中,affy会根据芯片平台自动化地从annotation中下载对应的cdf包,对于那些cdf文件没有收录在annotation中的芯片,就只能通过makecdfenv包手动创建对应的注释包了。
使用affy包读取cel文件的代码如下
library(affy)# 读取数据
data <- ReadAffy(celfile.path = "cel_file_dir")
核心就是ReadAffy函数,只需要提供cel文件所在文件夹的路径即可。
原始信号读取之后,我们需要将原始的探针水平的信号强度转变为基因水平的表达量,需要经过以下步骤
1. 读取探针水平的数据
2. 背景校正
3. 归一化
4. 探针特异性的背景校正,比如减去阴性对照的荧光强度
5. summary, 将一组探针的表达量合并为一个表达值水平
所有这些都通过一个函数expresso来执行,该函数非常灵活,包含了以下多个参数
1. bgcorrect.method
2. normalize.method
3. pmcorrect.method
4. summary.method
针对每一步骤都提供了很多的方法可供选择,展示如下
> library(affydata))> data(Dilution)
> normalize.methods(Dilution)
[1] "constant" "contrasts" "invariantset" "loess" "methods"
[6] "qspline" "quantiles" "quantiles.robust"
> bgcorrect.methods()
[1] "bg.correct" "mas" "none" "rma"
> pmcorrect.methods()
[1] "mas" "methods" "pmonly" "subtractmm"
> express.summary.stat.methods()
[1] "avgdiff" "liwong" "mas" "medianpolish" "playerout"
在expresso函数的基础上,封装了两个常见处理函数
1.mas5
2.rma
本质是固定了各种参数的值,从读取原始数据,到得到探针表达量的完整代码如下
library(affy)# 读取数据
data <- ReadAffy(celfile.path = "cel_file_dir")
# 归一化, 二选一
eset <- mas5(data)
est <- rma(data)
# 输出
write.exprs(eset, file="data.txt")
·end·

一个只分享干货的
生信公众号