计算测序深度是数据分析中的一个常规操作,常用的工具有以下几种
1. samtools depth
命令如下
samtools depth input.bam > sample.depth2. samtools mpileup
命令如下
samtools mpileup -A -Q 0 chrY.bam | cut -f1,2,4 > sample.depth3. bdetools genomecov
命令如下
bedtools genomecov -ibam input.bam -d > sample.depth尽管方法不同,但是输出结果的格式是相同的,示意如下
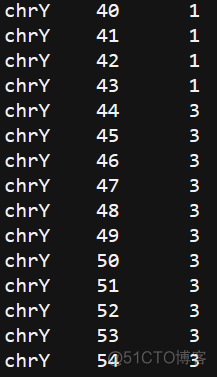
第一列是染色体名称,第二列是染色体上的坐标,第三列是对应的测序深度。原本以为计算测序深度就是这么轻松的一件事,但是在比较不同方法的输出结果时,却发现部分区域samtools计算的结果和bedtools的结果对应不上,结果如下
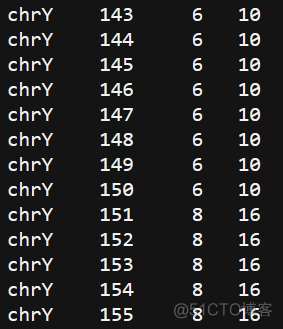
第三列为samtools软件计算出来的结果,第四列为bedtools软件计算出来的结果,可以看到,samtools的结果比bedtools少了很多。
为了确定这段区域真实的测序深度,将bam文件导入IGV软件之中进行查看,对应区域的结果如下
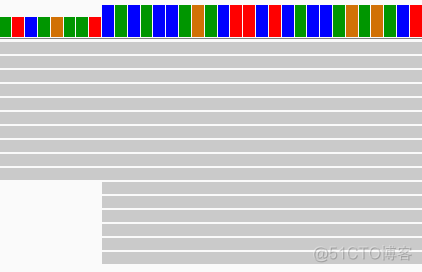
从reads来看,确实应该是10和16条,那么samtools计算出来的结果又是什么, 总不可能是samtools的bug吧,作为一个应用这个广泛的软件,不可能有如此低级的错误。
从结果来看,samtools在计算depth的过程中对reads进行了过滤,那么它过滤的原则是什么呢?通过查看bam文件中第二列的flag我找到了规律,143-150bp的reads有以下10条
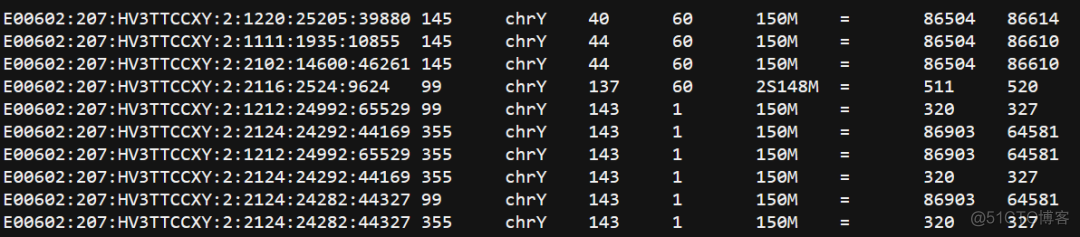
第二列的flag含义如下
0x91 145 PAIRED,REVERSE,READ20x163 355 PAIRED,PROPER_PAIR,MREVERSE,READ1,SECONDARY
0x63 99 PAIRED,PROPER_PAIR,MREVERSE,READ1
可以看到,其中有一个SECONDARY比对,对应数字355,这样的reads有4条,去除这4条之后,就是samtools计算的最终结果了。
作为SAM文件的官方操作工具,samtools内置了对flag的过滤, 而bedtools默认没有进行这样的过滤,只是简单统计了该区域比对上的reads数目。
在使用IGV展示测序深度时,可以用igvtools先计算出tdf文件再导入IGV中,这样比直接导入bam文件快很多。在tdf文件中,其测序深度和bedtools软件的计算结果是一致的,也是只统计了read数目,没有做过滤。
·end·

一个只分享干货的
生信公众号