bam文件记录了reads比对到参考基因组的详细信息,是NGS分析中的核心文件。该文件是一种二进制的格式,通常我们只能借助samtools这种特定的工具来转换成纯文本的格式进行查看,bam文件中包含的reads数量巨大,纯文本查看操作耗时且不够直观。
为了更好的查看序列比对,samtools也提功了tview这样的功能,来辅助查看序列比对。但是上述做法远没有基因组浏览器来的方便,将bam文件导入IGV之后,不仅可以灵活的选择想要查看的基因组区域,还能够查看测序深度的分布,exon的连接方式等更多的信息。
bam文件需要排序并建立索引之后才可以导入IGV, 这个前处理可以通过samtools来实现。对于bam文件,IGV提供了以下3种视图
1. alignment track
这部分展示的是序列的比对情况,通过缩放之后可以看到如下所示的结果
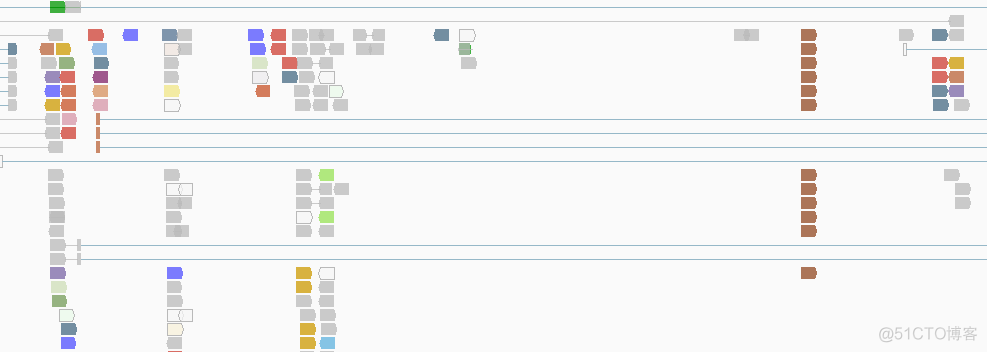
其中带箭头的矩形区域代表的就是测序的reads,矩形区域的箭头和比对到基因组的正负链有对应关系,比对到正链,箭头朝右,比对到负链,箭头朝左。
矩形的颜色也有着独特的含义,默认情况下,矩形的颜色为白色到灰色的渐变色,对应MAPQ值从小到大。对于双端测序而言,会出现R1和R2端比对到不同染色体的情况,此时根据另一端比对到的不同染色体赋予不同的颜色,对应的图例如下

矩形的颜色还可以反映很多的属性,比如基因组区域的插入和缺失,对于发生了缺失的基因组区域,用红色表示

对于发生了插入的基因组区域,用红色表示

也可以反映reads比对的方向和甲基化位点等,具体的颜色映射可以参考以下几个链接
http://software.broadinstitute.org/software/igv/interpreting_insert_sizehttp://software.broadinstitute.org/software/igv/interpreting_pair_orientations
http://software.broadinstitute.org/software/igv/interpreting_bisulfite_mode
将reads进一步放大之后,可以看到上面的碱基,示意如下

默认情况下,与参考基因组精确匹配的碱基用灰色表示,错配的碱基则根据A,T,C,G映射到不同的颜色。上述reads放大之后示意如下

同种类型的碱基对应同一个颜色,同时将碱基的质量值映射到透明度,所以可以看到,同种碱基的颜色透明度稍有不同。
在reads中,进一步对插入和缺失进行了标记,插入的碱基用Ⅰ表示,缺失用-表示,示意如下

2. coverage track
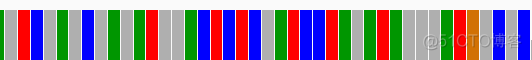
当缩放到一定程度时,会自动显示coverage track, 用柱状图展示每个位点的测序深度信息,示意如下
3. splice junction track
对于RNA_seq的数据,由于内含子的存在,reads比对到基因组上,是需要跳过内含子区的,splice junction track用曲线将外显子边界连接起来,示意如下

通过IGV, 可以直观的查看bam文件中的信息,即可以直接查看reads的详细比对情况,也可以查看测序深度等信息。
·end·

一个只分享干货的
生信公众号